Lee Edward B, Porta Sílvia, Michael Baer G, Xu Yan, Suh EunRan, Kwong Linda K, Elman Lauren, Grossman Murray, Lee Virginia M-Y, Irwin David J, Van Deerlin Vivianna M, Trojanowski John Q
Translational Neuropathology Research Laboratory, Department of Pathology and Laboratory Medicine, Perelman School of Medicine, University of Pennsylvania, 613A Stellar Chance Laboratories, 422 Curie Blvd, Philadelphia, PA, 19104, USA.
Center for Neurodegenerative Disease Research, University of Pennsylvania, Philadelphia, PA, USA.
Acta Neuropathol. 2017 Jul;134(1):65-78. doi: 10.1007/s00401-017-1679-9. Epub 2017 Jan 27.
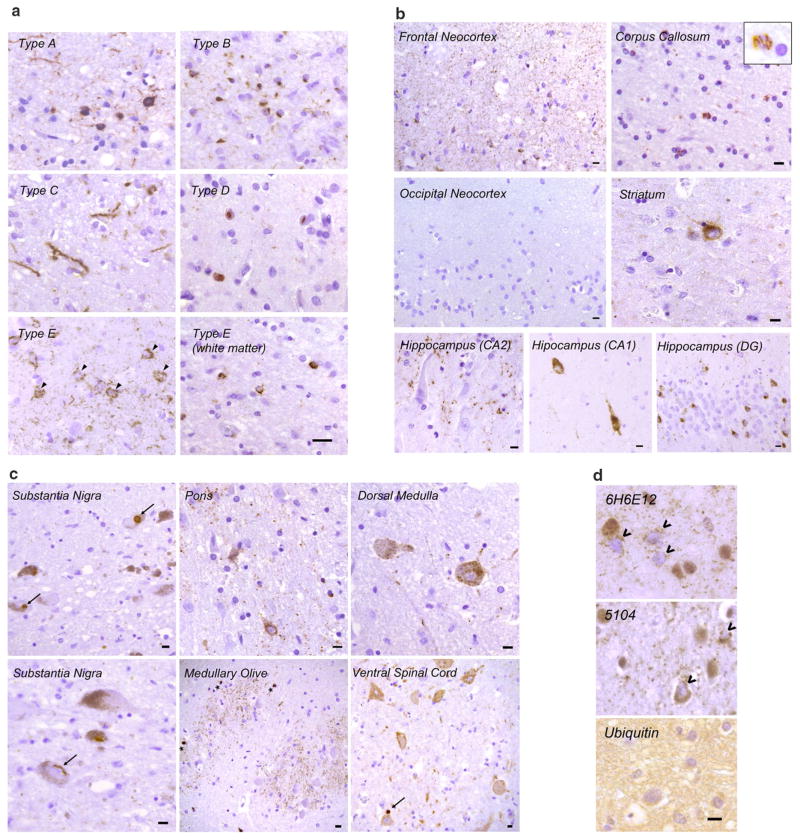
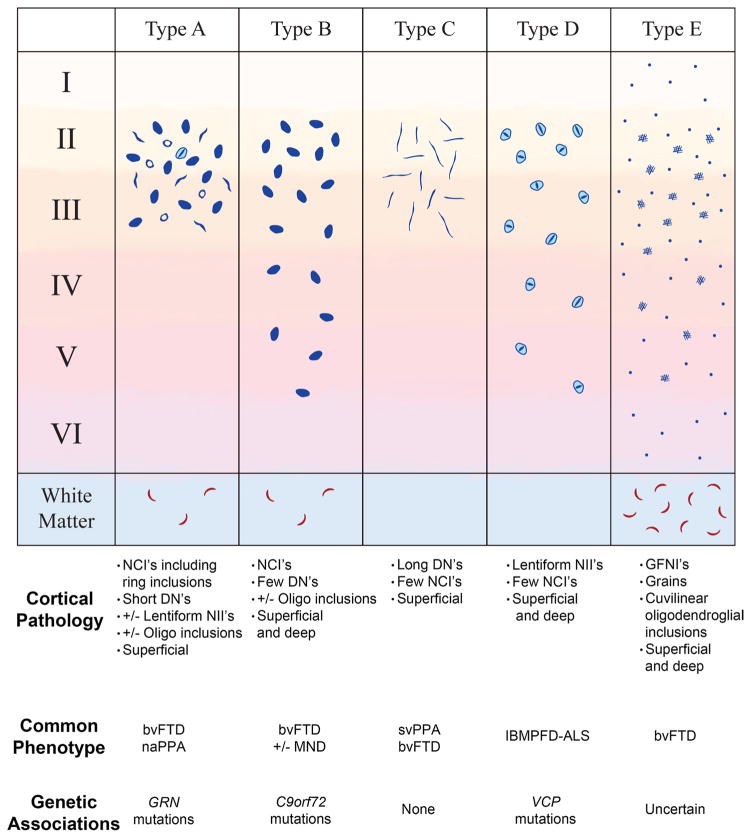
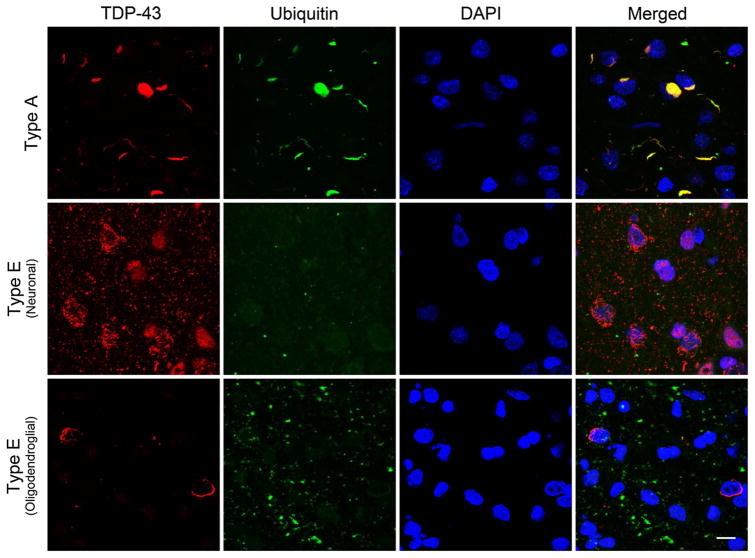
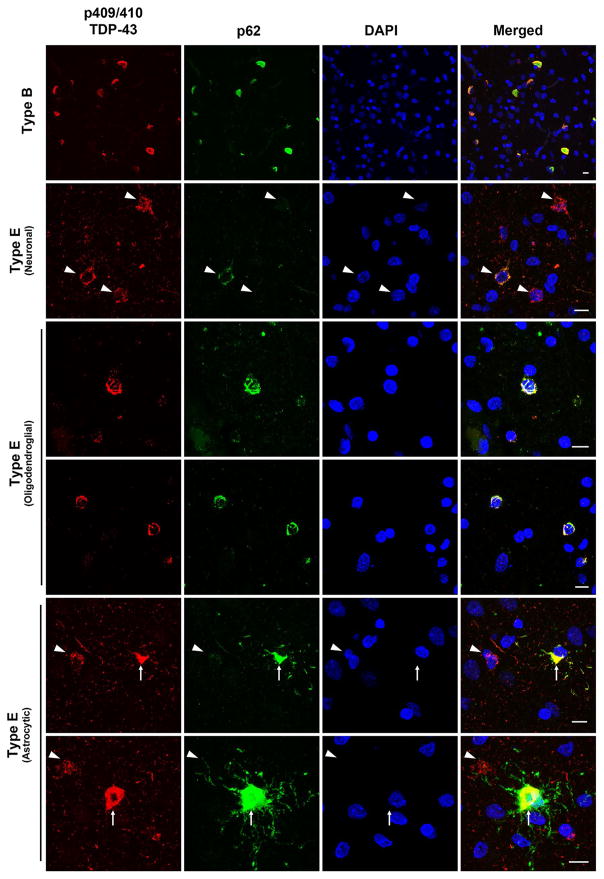
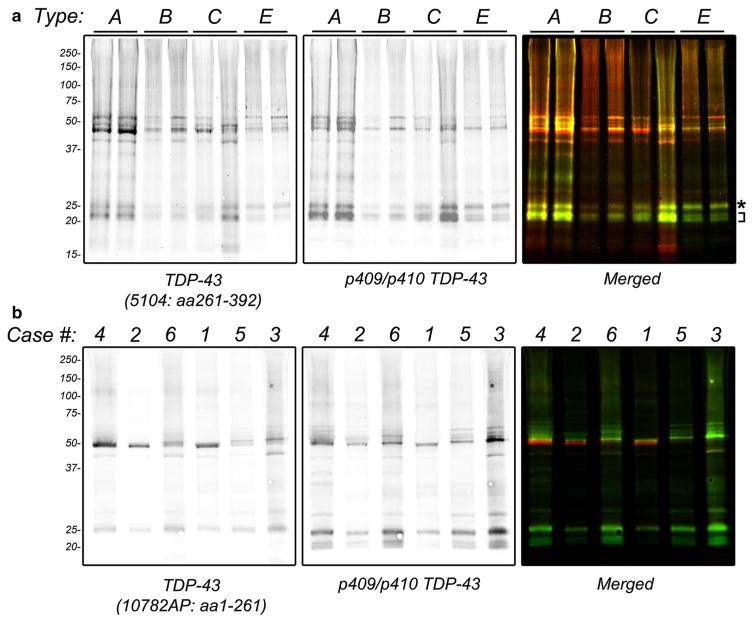
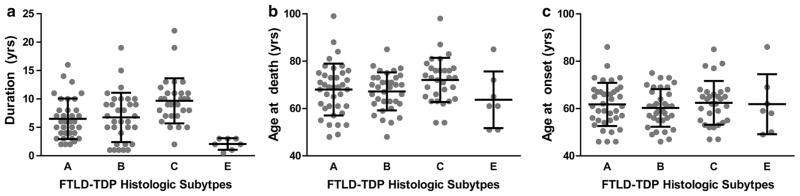
Frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) can typically be categorized into one of four distinct histopathologic patterns of TDP-43 pathology, types A to D. The strength of this histopathologic classification lies in the association between FTLD-TDP subtypes and various clinical and genetic features of disease. Seven cases of FTLD-TDP were identified here which were difficult to classify based on existing pathologic criteria. Distinct features common to these cases included TDP-43 aggregates over a wide neuroanatomic distribution comprised of granulofilamentous neuronal inclusions, abundant grains, and oligodendroglial inclusions. TDP-43 aggregates were phosphorylated and associated with loss of normal nuclear TDP-43 protein (nuclear clearance) but were negative for ubiquitin. Biochemical analysis confirmed the presence of insoluble and phosphorylated TDP-43 and also revealed a distinct pattern of TDP-43 C-terminal fragments relative to other FTLD-TDP subtypes. Finally, these cases were uniformly associated with a very rapid clinical course culminating in death within ~3 years of disease onset. We suggest that these cases may represent a unique clinicopathologic subtype of FTLD-TDP which we provisionally call "type E." The immature appearance of TDP-43 aggregates, widespread distribution, uniform biochemical profile and rapid clinical course highlights the clinical and pathologic variability within FTLD-TDP, and raises the possibility that type E neuropathology is the sequelae of a particularly virulent strain of TDP-43 proteinopathy.
伴有TDP-43包涵体的额颞叶变性(FTLD-TDP)通常可分为TDP-43病理的四种不同组织病理学模式之一,即A至D型。这种组织病理学分类的优势在于FTLD-TDP亚型与疾病的各种临床和遗传特征之间的关联。在此确定了7例FTLD-TDP病例,根据现有的病理学标准难以进行分类。这些病例的共同显著特征包括TDP-43聚集体在广泛的神经解剖分布中,由颗粒丝状神经元包涵体、大量颗粒和少突胶质细胞包涵体组成。TDP-43聚集体发生磷酸化,与正常核TDP-43蛋白的缺失(核清除)相关,但泛素检测为阴性。生化分析证实存在不溶性和磷酸化的TDP-43,并且还揭示了相对于其他FTLD-TDP亚型的TDP-43 C末端片段的独特模式。最后,这些病例均与非常快速的临床病程相关,在疾病发作后约3年内死亡。我们认为这些病例可能代表了FTLD-TDP的一种独特的临床病理亚型,我们暂时将其称为“E型”。TDP-43聚集体的不成熟外观、广泛分布、一致的生化特征和快速的临床病程突出了FTLD-TDP内的临床和病理变异性,并增加了E型神经病理学是一种特别恶性的TDP-43蛋白病毒株后遗症的可能性。