Department of Cell and Developmental Biology, University College London, Gower Street, London WC1E 6BT, United Kingdom.
Department of Cell and Developmental Biology, University College London, Gower Street, London WC1E 6BT, United Kingdom; Department of Biomedical Sciences, University of Padua, 35131 Padua, Italy.
Biochim Biophys Acta Mol Cell Res. 2017 Jun;1864(6):1009-1017. doi: 10.1016/j.bbamcr.2017.01.015. Epub 2017 Jan 26.
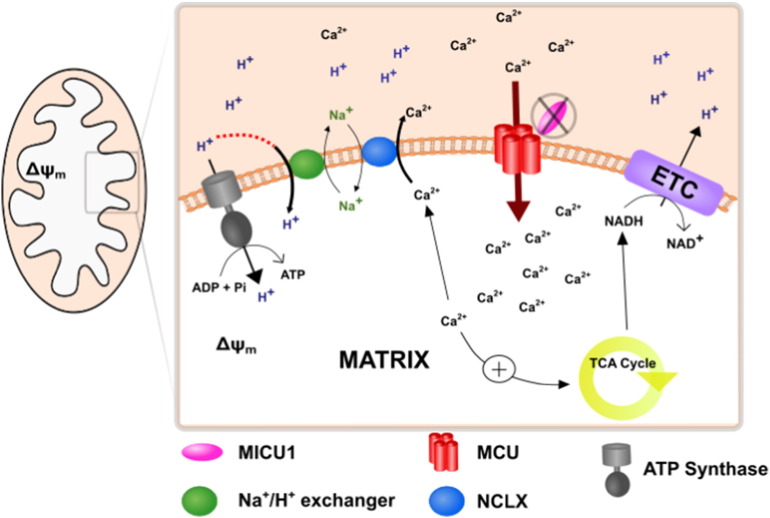
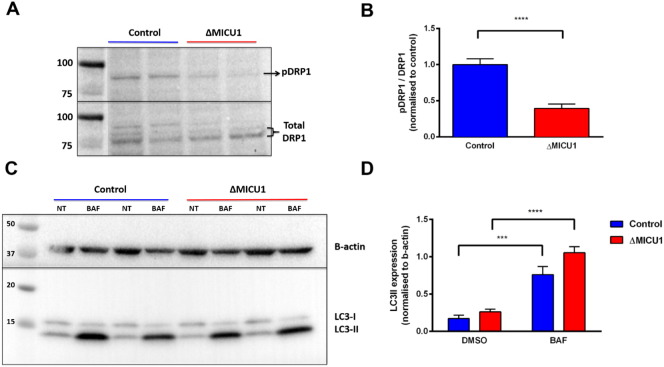
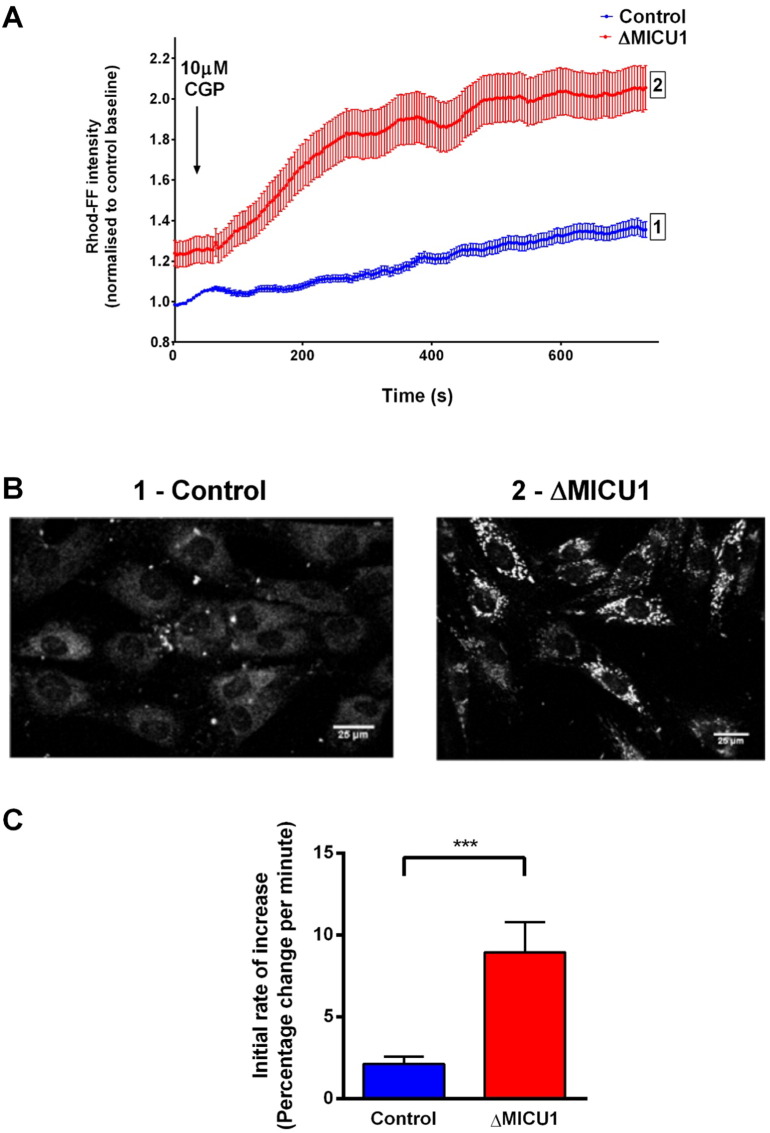
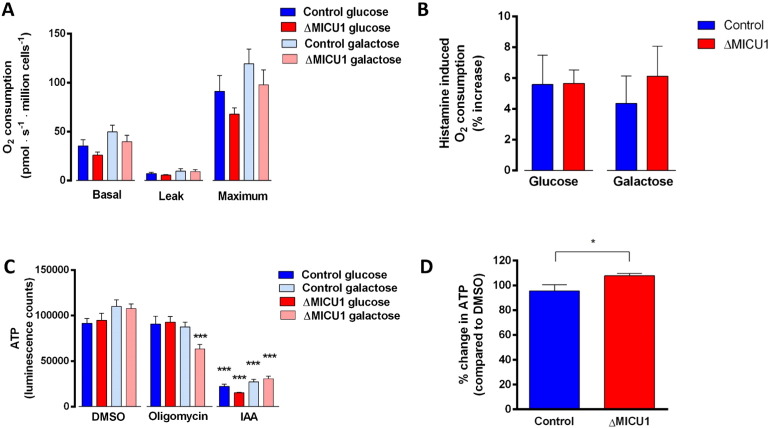
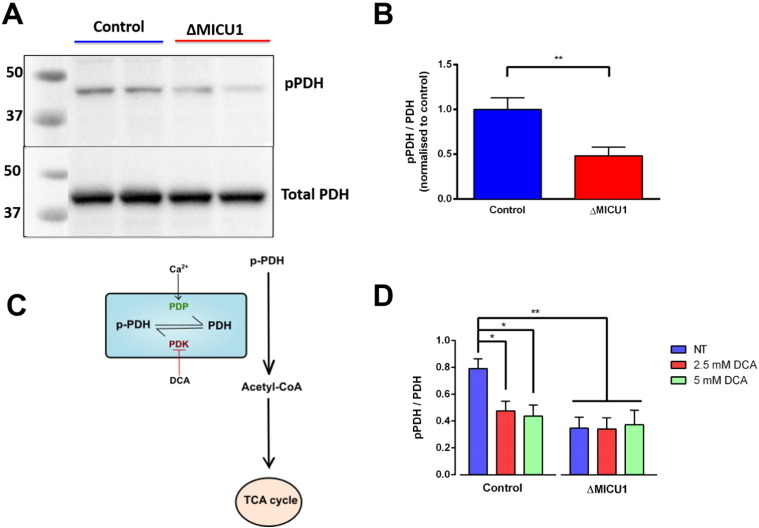
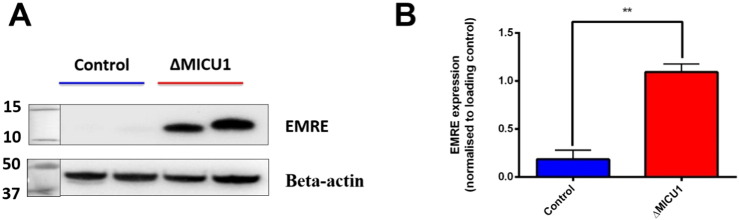
Loss of function mutations of the protein MICU1, a regulator of mitochondrial Ca uptake, cause a neuronal and muscular disorder characterised by impaired cognition, muscle weakness and an extrapyramidal motor disorder. We have shown previously that MICU1 mutations cause increased resting mitochondrial Ca concentration ([Ca]). We now explore the functional consequences of MICU1 mutations in patient derived fibroblasts in order to clarify the underlying pathophysiology of this disorder. We propose that deregulation of mitochondrial Ca uptake through loss of MICU1 raises resting [Ca], initiating a futile Ca cycle, whereby continuous mitochondrial Ca influx is balanced by Ca efflux through the sodium calcium exchanger (NLCX). Thus, inhibition of NCLX by CGP-37157 caused rapid mitochondrial Ca accumulation in patient but not control cells. We suggest that increased NCLX activity will increase sodium/proton exchange, potentially undermining oxidative phosphorylation, although this is balanced by dephosphorylation and activation of pyruvate dehydrogenase (PDH) in response to the increased [Ca]. Consistent with this model, while ATP content in patient derived or control fibroblasts was not different, ATP increased significantly in response to CGP-37157 in the patient but not the control cells. In addition, EMRE expression levels were altered in MICU1 patient cells compared to the controls. The MICU1 mutations were associated with mitochondrial fragmentation which we show is related to altered DRP1 phosphorylation. Thus, MICU1 serves as a signal-noise discriminator in mitochondrial calcium signalling, limiting the energetic costs of mitochondrial Ca signalling which may undermine oxidative phosphorylation, especially in tissues with highly dynamic energetic demands. This article is part of a Special Issue entitled: ECS Meeting edited by Claus Heizmann, Joachim Krebs and Jacques Haiech.
MICU1 蛋白是线粒体钙摄取的调节因子,其功能丧失突变可导致以认知障碍、肌肉无力和锥体外系运动障碍为特征的神经元和肌肉疾病。我们之前已经表明,MICU1 突变会导致静息线粒体钙浓度 ([Ca]) 升高。我们现在探索 MICU1 突变在患者来源的成纤维细胞中的功能后果,以阐明这种疾病的潜在病理生理学。我们提出,通过 MICU1 的缺失导致线粒体钙摄取的失调会升高静息 [Ca],引发无效的钙循环,从而连续的线粒体钙内流通过钠钙交换器 (NLCX) 平衡钙外排。因此,CGP-37157 抑制 NCLX 会导致患者而非对照细胞中快速的线粒体钙积累。我们认为,增加的 NCLX 活性将增加钠/质子交换,可能破坏氧化磷酸化,尽管这通过增加 [Ca] 时磷酸化和丙酮酸脱氢酶 (PDH) 的激活来平衡。与该模型一致,尽管患者来源的或对照的成纤维细胞中的 ATP 含量没有差异,但 CGP-37157 会显著增加患者而不是对照细胞中的 ATP。此外,与对照细胞相比,MICU1 患者细胞中的 EMRE 表达水平发生改变。MICU1 突变与线粒体片段化有关,我们发现这与 DRP1 磷酸化的改变有关。因此,MICU1 作为线粒体钙信号中的信号噪声甄别器,限制线粒体钙信号的能量成本,这可能破坏氧化磷酸化,尤其是在具有高度动态能量需求的组织中。本文是由 Claus Heizmann、Joachim Krebs 和 Jacques Haiech 编辑的特刊题为:ECS Meeting 的一部分。