Zhu Xun, Ching Travers, Pan Xinghua, Weissman Sherman M, Garmire Lana
Epidemiology Program, University of Hawaii Cancer Center, Honolulu, HI, United States; Molecular Biosciences and Bioengineering Graduate Program, University of Hawaii at Manoa, Honolulu, United States.
Department of Genetics, Yale University , New Haven , CT , United States.
PeerJ. 2017 Jan 19;5:e2888. doi: 10.7717/peerj.2888. eCollection 2017.
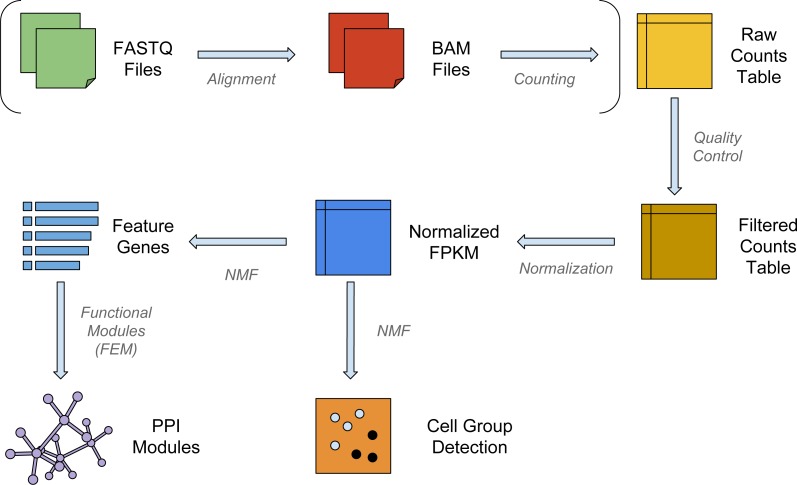
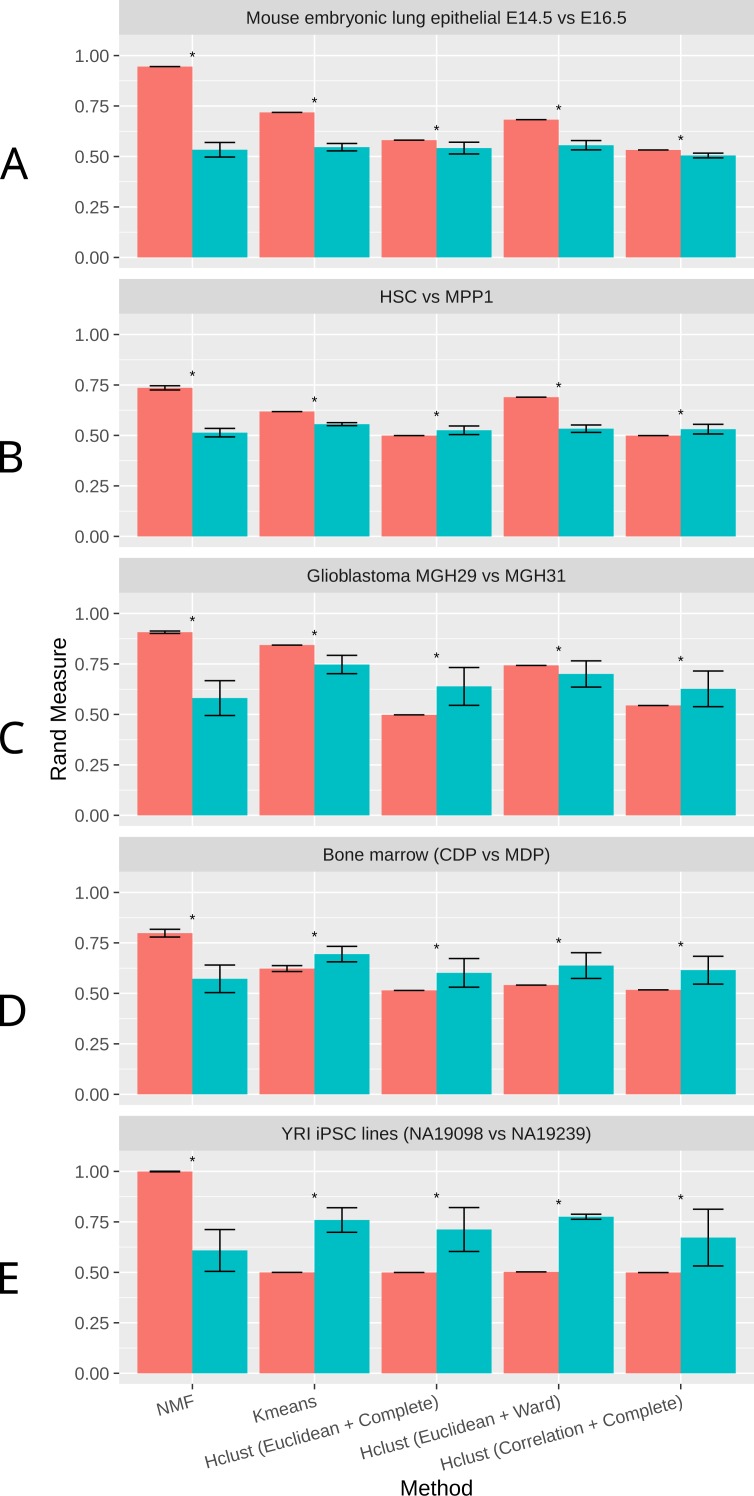
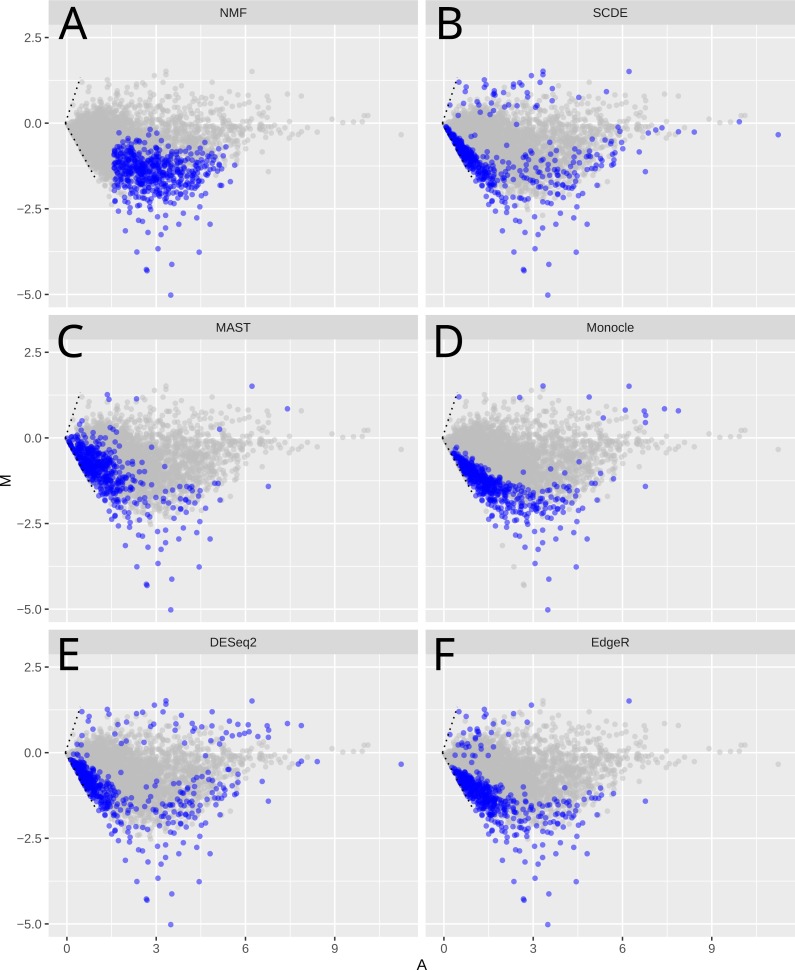
Single-cell RNA-Sequencing (scRNA-Seq) is a fast-evolving technology that enables the understanding of biological processes at an unprecedentedly high resolution. However, well-suited bioinformatics tools to analyze the data generated from this new technology are still lacking. Here we investigate the performance of non-negative matrix factorization (NMF) method to analyze a wide variety of scRNA-Seq datasets, ranging from mouse hematopoietic stem cells to human glioblastoma data. In comparison to other unsupervised clustering methods including K-means and hierarchical clustering, NMF has higher accuracy in separating similar groups in various datasets. We ranked genes by their importance scores (-scores) in separating these groups, and discovered that NMF uniquely identifies genes expressed at intermediate levels as top-ranked genes. Finally, we show that in conjugation with the modularity detection method FEM, NMF reveals meaningful protein-protein interaction modules. In summary, we propose that NMF is a desirable method to analyze heterogeneous single-cell RNA-Seq data. The NMF based subpopulation detection package is available at: https://github.com/lanagarmire/NMFEM.
单细胞RNA测序(scRNA-Seq)是一项快速发展的技术,能够以前所未有的高分辨率理解生物过程。然而,仍缺乏适用于分析这项新技术所产生数据的生物信息学工具。在此,我们研究非负矩阵分解(NMF)方法在分析各种scRNA-Seq数据集时的性能,这些数据集涵盖从小鼠造血干细胞到人类胶质母细胞瘤数据等。与包括K均值和层次聚类在内的其他无监督聚类方法相比,NMF在分离各种数据集中的相似组时具有更高的准确性。我们根据基因在分离这些组中的重要性得分(-得分)对基因进行排序,发现NMF独特地将中等水平表达的基因识别为排名靠前的基因。最后,我们表明,与模块性检测方法FEM结合时,NMF能揭示有意义的蛋白质-蛋白质相互作用模块。总之,我们提出NMF是分析异质单细胞RNA-Seq数据的理想方法。基于NMF的亚群检测软件包可在以下网址获取:https://github.com/lanagarmire/NMFEM 。