Jansen Iris E, Ye Hui, Heetveld Sasja, Lechler Marie C, Michels Helen, Seinstra Renée I, Lubbe Steven J, Drouet Valérie, Lesage Suzanne, Majounie Elisa, Gibbs J Raphael, Nalls Mike A, Ryten Mina, Botia Juan A, Vandrovcova Jana, Simon-Sanchez Javier, Castillo-Lizardo Melissa, Rizzu Patrizia, Blauwendraat Cornelis, Chouhan Amit K, Li Yarong, Yogi Puja, Amin Najaf, van Duijn Cornelia M, Morris Huw R, Brice Alexis, Singleton Andrew B, David Della C, Nollen Ellen A, Jain Shushant, Shulman Joshua M, Heutink Peter
German Center for Neurodegenerative Diseases (DZNE), Otfried-Müller-Str. 23, Tübingen, 72076, Germany.
Department of Clinical Genetics, VU University Medical Center, Amsterdam, 1081HZ, The Netherlands.
Genome Biol. 2017 Jan 30;18(1):22. doi: 10.1186/s13059-017-1147-9.
Whole-exome sequencing (WES) has been successful in identifying genes that cause familial Parkinson's disease (PD). However, until now this approach has not been deployed to study large cohorts of unrelated participants. To discover rare PD susceptibility variants, we performed WES in 1148 unrelated cases and 503 control participants. Candidate genes were subsequently validated for functions relevant to PD based on parallel RNA-interference (RNAi) screens in human cell culture and Drosophila and C. elegans models.
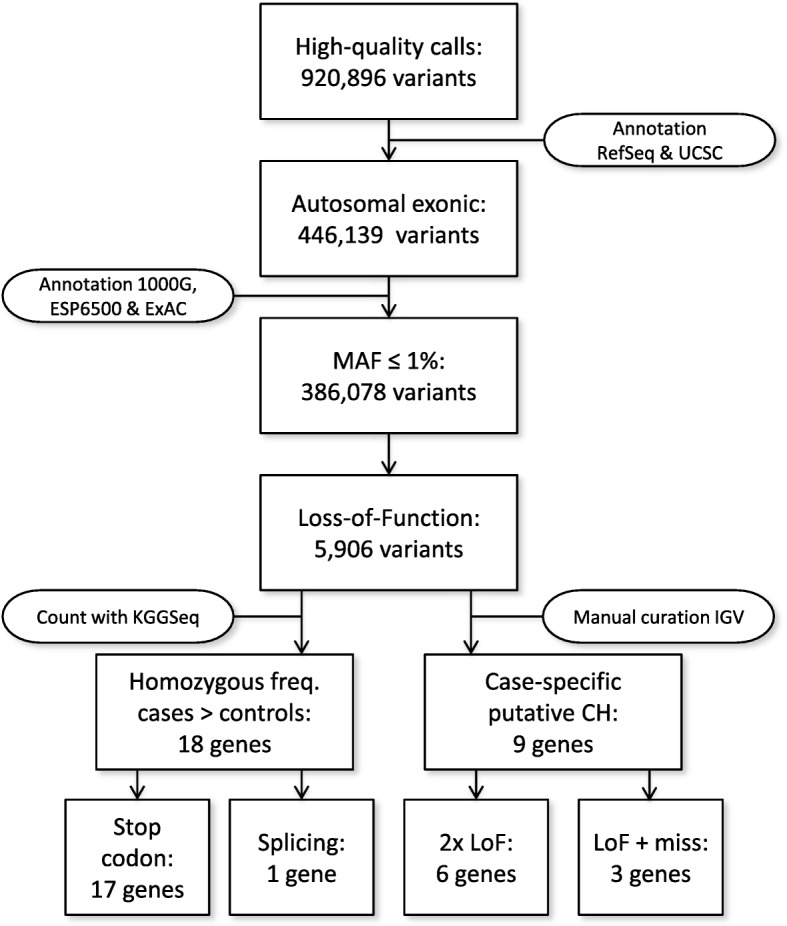
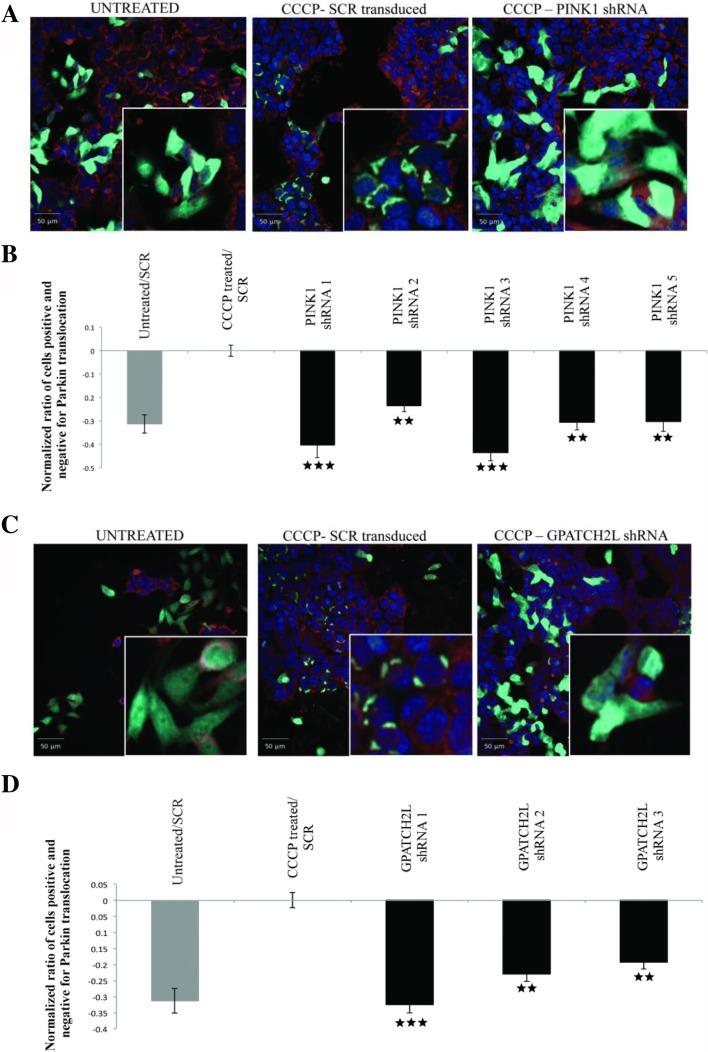
Assuming autosomal recessive inheritance, we identify 27 genes that have homozygous or compound heterozygous loss-of-function variants in PD cases. Definitive replication and confirmation of these findings were hindered by potential heterogeneity and by the rarity of the implicated alleles. We therefore looked for potential genetic interactions with established PD mechanisms. Following RNAi-mediated knockdown, 15 of the genes modulated mitochondrial dynamics in human neuronal cultures and four candidates enhanced α-synuclein-induced neurodegeneration in Drosophila. Based on complementary analyses in independent human datasets, five functionally validated genes-GPATCH2L, UHRF1BP1L, PTPRH, ARSB, and VPS13C-also showed evidence consistent with genetic replication.
By integrating human genetic and functional evidence, we identify several PD susceptibility gene candidates for further investigation. Our approach highlights a powerful experimental strategy with broad applicability for future studies of disorders with complex genetic etiologies.
全外显子组测序(WES)已成功鉴定出导致家族性帕金森病(PD)的基因。然而,到目前为止,这种方法尚未应用于研究大量无血缘关系的参与者队列。为了发现罕见的PD易感性变异,我们对1148例无血缘关系的病例和503名对照参与者进行了WES。随后,基于在人类细胞培养以及果蝇和秀丽隐杆线虫模型中的平行RNA干扰(RNAi)筛选,对候选基因与PD相关的功能进行了验证。
假设常染色体隐性遗传,我们在PD病例中鉴定出27个具有纯合或复合杂合功能丧失变异的基因。潜在的异质性和所涉及等位基因的稀有性阻碍了这些发现的明确复制和确认。因此,我们寻找与既定PD机制的潜在遗传相互作用。在RNAi介导的敲低后,其中15个基因在人类神经元培养物中调节线粒体动力学,4个候选基因在果蝇中增强了α-突触核蛋白诱导的神经退行性变。基于在独立人类数据集中的补充分析,5个功能验证基因——GPATCH2L、UHRF1BP1L、PTPRH、ARSB和VPS13C——也显示出与基因复制一致的证据。
通过整合人类遗传和功能证据,我们鉴定出几个PD易感性候选基因以供进一步研究。我们的方法突出了一种强大的实验策略,对未来复杂遗传病因疾病的研究具有广泛适用性。