Jiao Wen-Biao, Accinelli Gonzalo Garcia, Hartwig Benjamin, Kiefer Christiane, Baker David, Severing Edouard, Willing Eva-Maria, Piednoel Mathieu, Woetzel Stefan, Madrid-Herrero Eva, Huettel Bruno, Hümann Ulrike, Reinhard Richard, Koch Marcus A, Swan Daniel, Clavijo Bernardo, Coupland George, Schneeberger Korbinian
Department of Plant Developmental Biology, Max Planck Institute for Plant Breeding Research, 50829 Cologne, Germany.
Earlham Institute, Norwich Research Park, Norwich NR4 7UH, United Kingdom.
Genome Res. 2017 May;27(5):778-786. doi: 10.1101/gr.213652.116. Epub 2017 Feb 3.
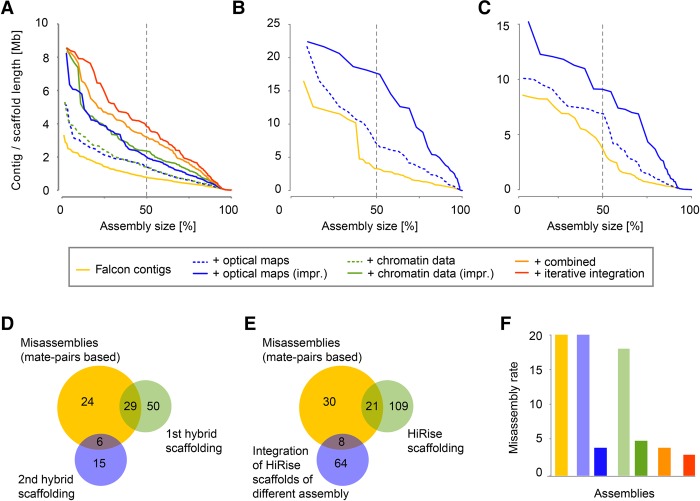
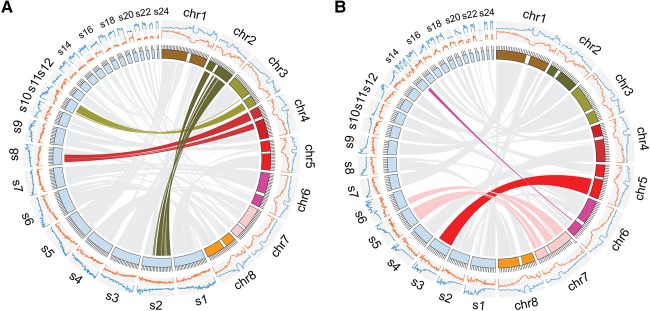
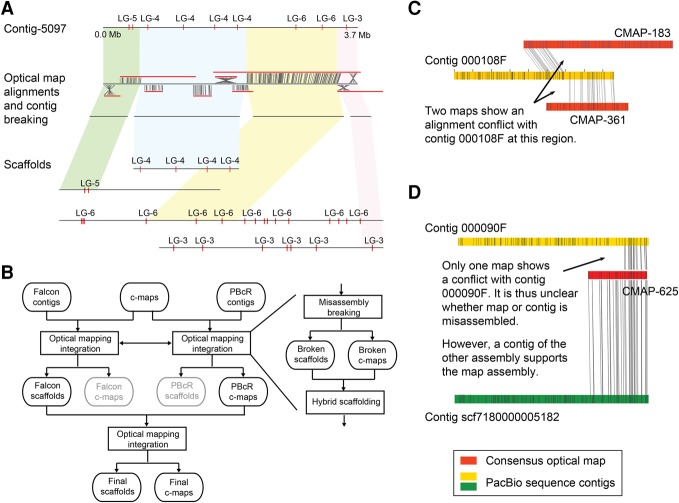
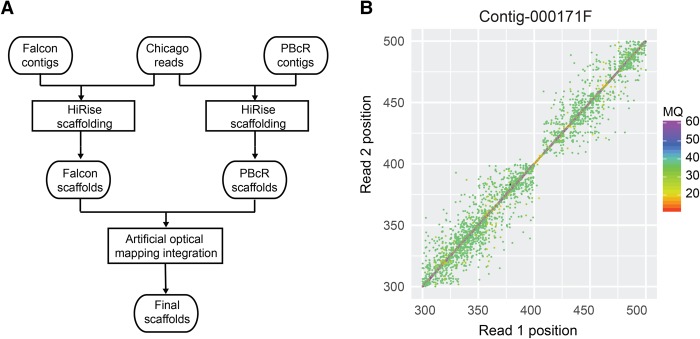
Long-read sequencing can overcome the weaknesses of short reads in the assembly of eukaryotic genomes; however, at present additional scaffolding is needed to achieve chromosome-level assemblies. We generated Pacific Biosciences (PacBio) long-read data of the genomes of three relatives of the model plant and assembled all three genomes into only a few hundred contigs. To improve the contiguities of these assemblies, we generated BioNano Genomics optical mapping and Dovetail Genomics chromosome conformation capture data for genome scaffolding. Despite their technical differences, optical mapping and chromosome conformation capture performed similarly and doubled N50 values. After improving both integration methods, assembly contiguity reached chromosome-arm-levels. We rigorously assessed the quality of contigs and scaffolds using Illumina mate-pair libraries and genetic map information. This showed that PacBio assemblies have high sequence accuracy but can contain several misassemblies, which join unlinked regions of the genome. Most, but not all, of these misjoints were removed during the integration of the optical mapping and chromosome conformation capture data. Even though none of the centromeres were fully assembled, the scaffolds revealed large parts of some centromeric regions, even including some of the heterochromatic regions, which are not present in gold standard reference sequences.
长读长测序可以克服短读长在真核生物基因组组装中的弱点;然而,目前需要额外的支架构建来实现染色体水平的组装。我们生成了模式植物三个近缘种基因组的太平洋生物科学公司(PacBio)长读长数据,并将这三个基因组组装成仅几百个重叠群。为了提高这些组装的连续性,我们生成了BioNano Genomics光学图谱和Dovetail Genomics染色体构象捕获数据用于基因组支架构建。尽管它们在技术上存在差异,但光学图谱和染色体构象捕获的表现相似,N50值翻倍。在改进了两种整合方法后,组装的连续性达到了染色体臂水平。我们使用Illumina配对文库和遗传图谱信息严格评估了重叠群和支架的质量。这表明PacBio组装具有较高的序列准确性,但可能包含一些错配,这些错配连接了基因组中不相连的区域。在光学图谱和染色体构象捕获数据整合过程中,大部分(但不是全部)这些错配被消除。尽管没有一个着丝粒被完全组装,但支架揭示了一些着丝粒区域的大部分,甚至包括一些在金标准参考序列中不存在的异染色质区域。