Michael Todd P, Jupe Florian, Bemm Felix, Motley S Timothy, Sandoval Justin P, Lanz Christa, Loudet Olivier, Weigel Detlef, Ecker Joseph R
J. Craig Venter Institute, La Jolla, CA, 92037, USA.
Genomic Analysis Laboratory, The Salk Institute for Biological Studies, La Jolla, CA, 92037, USA.
Nat Commun. 2018 Feb 7;9(1):541. doi: 10.1038/s41467-018-03016-2.
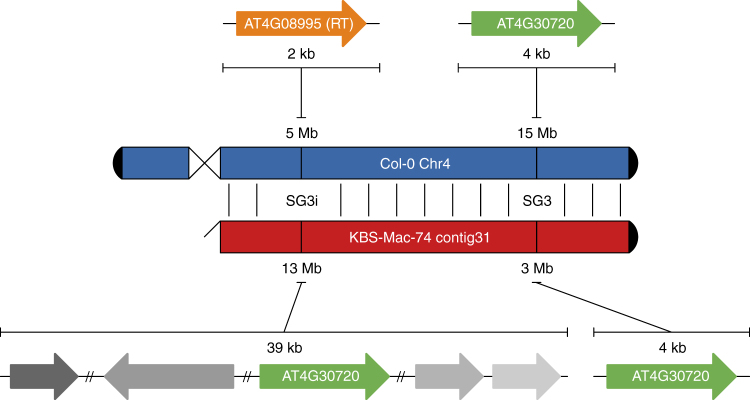
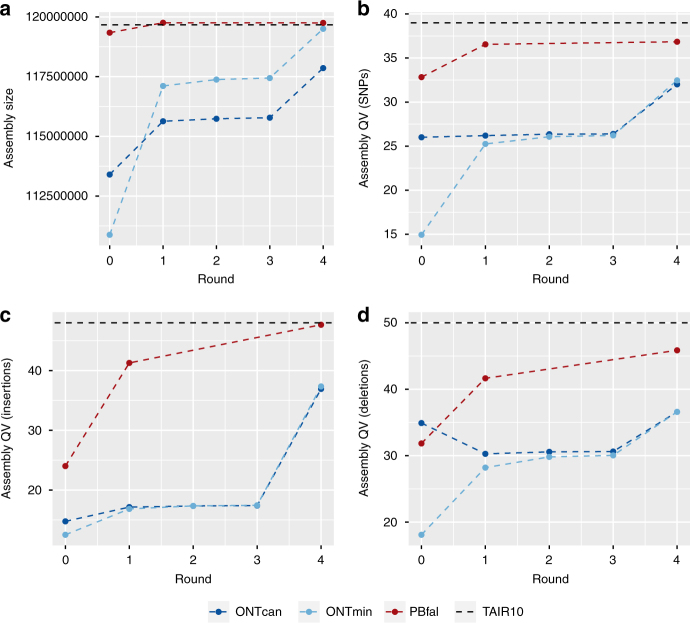
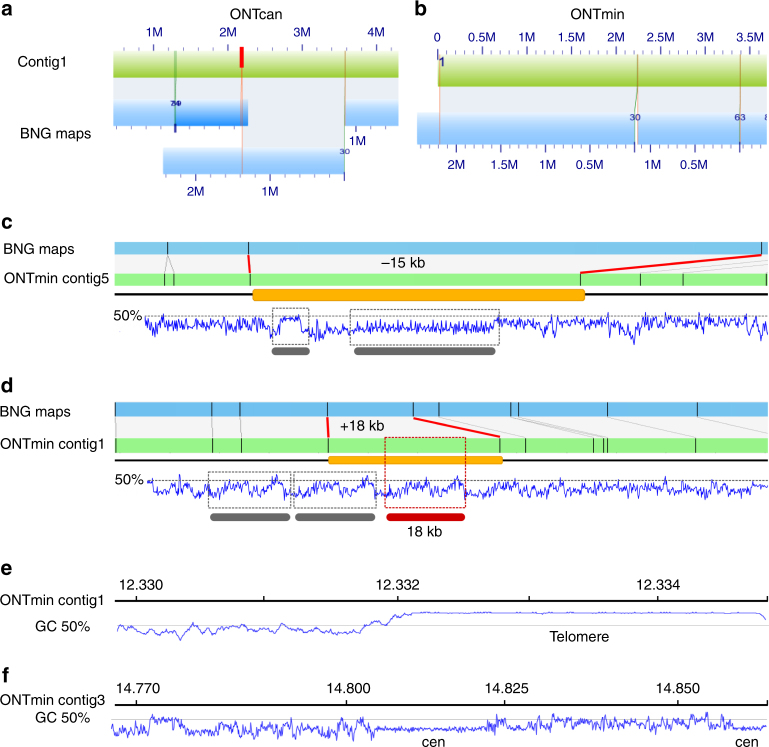
The handheld Oxford Nanopore MinION sequencer generates ultra-long reads with minimal cost and time requirements, which makes sequencing genomes at the bench feasible. Here, we sequence the gold standard Arabidopsis thaliana genome (KBS-Mac-74 accession) on the bench with the MinION sequencer, and assemble the genome using typical consumer computing hardware (4 Cores, 16 Gb RAM) into chromosome arms (62 contigs with an N50 length of 12.3 Mb). We validate the contiguity and quality of the assembly with two independent single-molecule technologies, Bionano optical genome maps and Pacific Biosciences Sequel sequencing. The new A. thaliana KBS-Mac-74 genome enables resolution of a quantitative trait locus that had previously been recalcitrant to a Sanger-based BAC sequencing approach. In summary, we demonstrate that even when the purpose is to understand complex structural variation at a single region of the genome, complete genome assembly is becoming the simplest way to achieve this goal.
手持式牛津纳米孔MinION测序仪能够以最低的成本和时间要求生成超长读长,这使得在实验台上对基因组进行测序成为可能。在这里,我们使用MinION测序仪在实验台上对黄金标准的拟南芥基因组(KBS-Mac-74 accession)进行测序,并使用典型的消费级计算硬件(4核,16GB内存)将基因组组装成染色体臂(62个重叠群,N50长度为12.3Mb)。我们使用两种独立的单分子技术,即Bionano光学基因组图谱和太平洋生物科学公司的Sequel测序,验证了组装的连续性和质量。新的拟南芥KBS-Mac-74基因组能够解析一个以前难以用基于桑格法的BAC测序方法解决的数量性状位点。总之,我们证明,即使目的是了解基因组单个区域的复杂结构变异,完整的基因组组装也正成为实现这一目标的最简单方法。