Truong Duy Tin, Tett Adrian, Pasolli Edoardo, Huttenhower Curtis, Segata Nicola
Centre for Integrative Biology, University of Trento, 38123 Trento, Italy.
Biostatistics Department, Harvard School of Public Health, Boston, Massachusetts 02115, USA.
Genome Res. 2017 Apr;27(4):626-638. doi: 10.1101/gr.216242.116. Epub 2017 Feb 6.
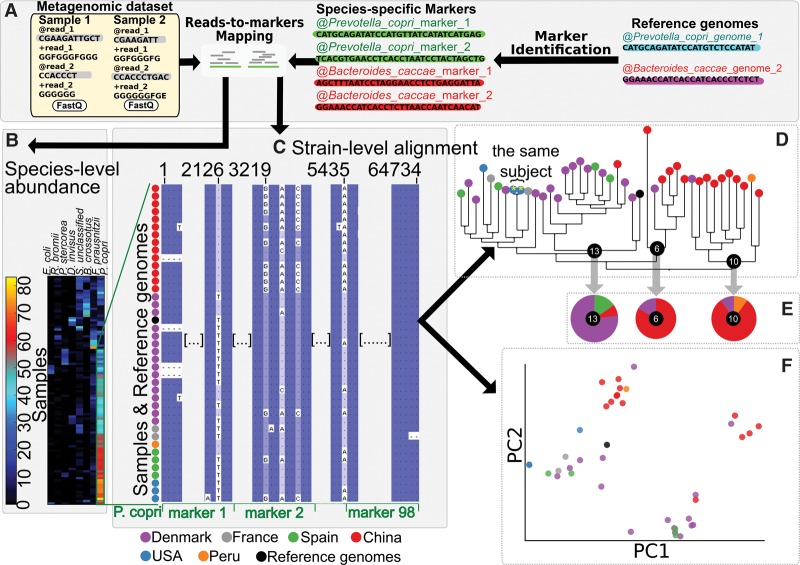
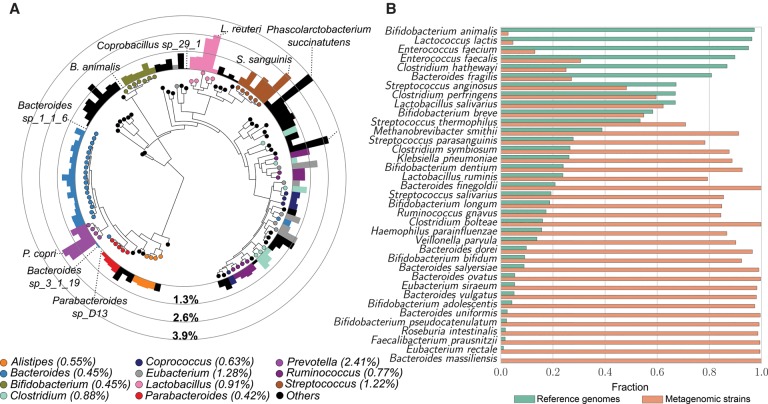
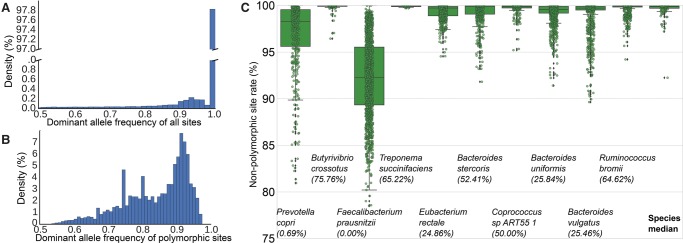
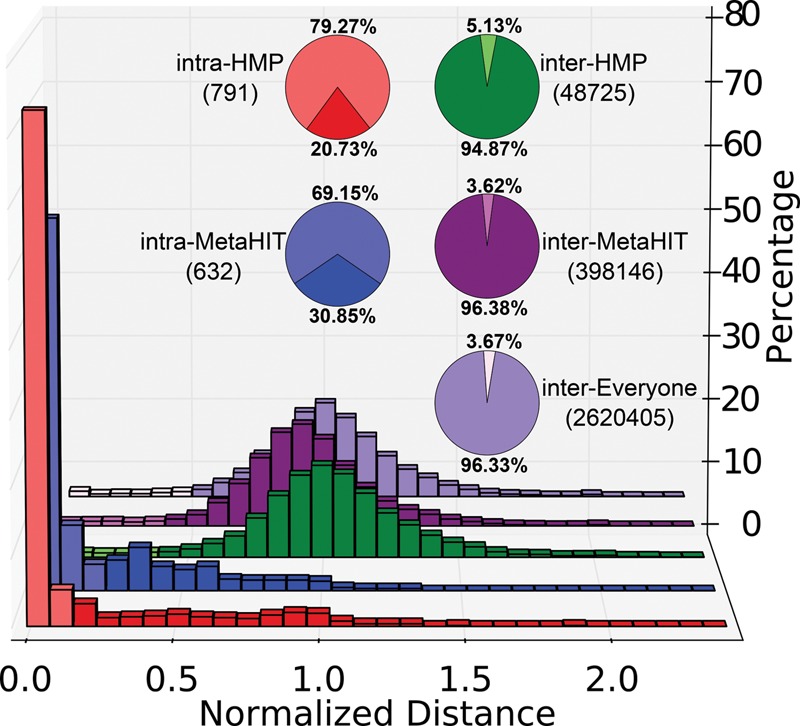
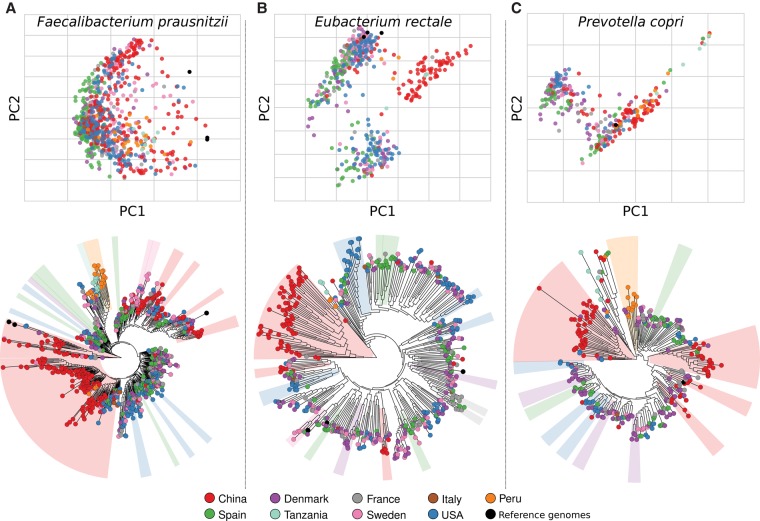
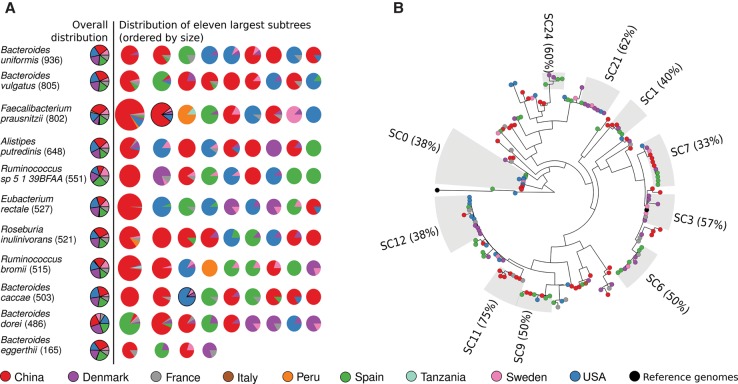
Among the human health conditions linked to microbial communities, phenotypes are often associated with only a subset of strains within causal microbial groups. Although it has been critical for decades in microbial physiology to characterize individual strains, this has been challenging when using culture-independent high-throughput metagenomics. We introduce StrainPhlAn, a novel metagenomic strain identification approach, and apply it to characterize the genetic structure of thousands of strains from more than 125 species in more than 1500 gut metagenomes drawn from populations spanning North and South American, European, Asian, and African countries. The method relies on per-sample dominant sequence variant reconstruction within species-specific marker genes. It identified primarily subject-specific strain variants (<5% inter-subject strain sharing), and we determined that a single strain typically dominated each species and was retained over time (for >70% of species). Microbial population structure was correlated in several distinct ways with the geographic structure of the host population. In some cases, discrete subspecies (e.g., for and ) or continuous microbial genetic variations (e.g., for ) were associated with geographically distinct human populations, whereas few strains occurred in multiple unrelated cohorts. We further estimated the genetic variability of gut microbes, with species appearing remarkably consistent (0.45% median number of nucleotide variants between strains), whereas was among the most plastic gut colonizers. We thus characterize here the population genetics of previously inaccessible intestinal microbes, providing a comprehensive strain-level genetic overview of the gut microbial diversity.
在与微生物群落相关的人类健康状况中,表型通常仅与致病微生物群体中的一部分菌株相关。尽管几十年来在微生物生理学中鉴定单个菌株至关重要,但在使用非培养高通量宏基因组学时,这一直具有挑战性。我们引入了StrainPhlAn,一种新型的宏基因组菌株鉴定方法,并将其应用于表征来自北美、南美、欧洲、亚洲和非洲国家人群的1500多个肠道宏基因组中125多个物种的数千个菌株的遗传结构。该方法依赖于物种特异性标记基因内每个样本的优势序列变异重建。它主要鉴定出个体特异性菌株变异(个体间菌株共享率<5%),并且我们确定单个菌株通常在每个物种中占主导地位并随时间保持(>70%的物种)。微生物种群结构与宿主种群的地理结构以几种不同方式相关。在某些情况下,离散的亚种(例如,对于 和 )或连续的微生物遗传变异(例如,对于 )与地理上不同的人类群体相关,而很少有菌株出现在多个不相关的队列中。我们进一步估计了肠道微生物的遗传变异性, 物种表现出显著的一致性(菌株间核苷酸变异中位数为0.45%),而 是肠道定植菌中可塑性最强的之一。因此,我们在此表征了以前难以研究的肠道微生物的群体遗传学,提供了肠道微生物多样性的全面菌株水平遗传概述。