Compagno Mara, Wang Qi, Pighi Chiara, Cheong Taek-Chin, Meng Fei-Long, Poggio Teresa, Yeap Leng-Siew, Karaca Elif, Blasco Rafael B, Langellotto Fernanda, Ambrogio Chiara, Voena Claudia, Wiestner Adrian, Kasar Siddha N, Brown Jennifer R, Sun Jing, Wu Catherine J, Gostissa Monica, Alt Frederick W, Chiarle Roberto
Department of Pathology, Children's Hospital Boston and Harvard Medical School, Boston, Massachusetts 02115, USA.
Howard Hughes Medical Institute, Program in Cellular and Molecular Medicine, Boston Children's Hospital, and Department of Genetics, Harvard Medical School, Boston, Massachusetts 02115, USA.
Nature. 2017 Feb 23;542(7642):489-493. doi: 10.1038/nature21406. Epub 2017 Feb 15.
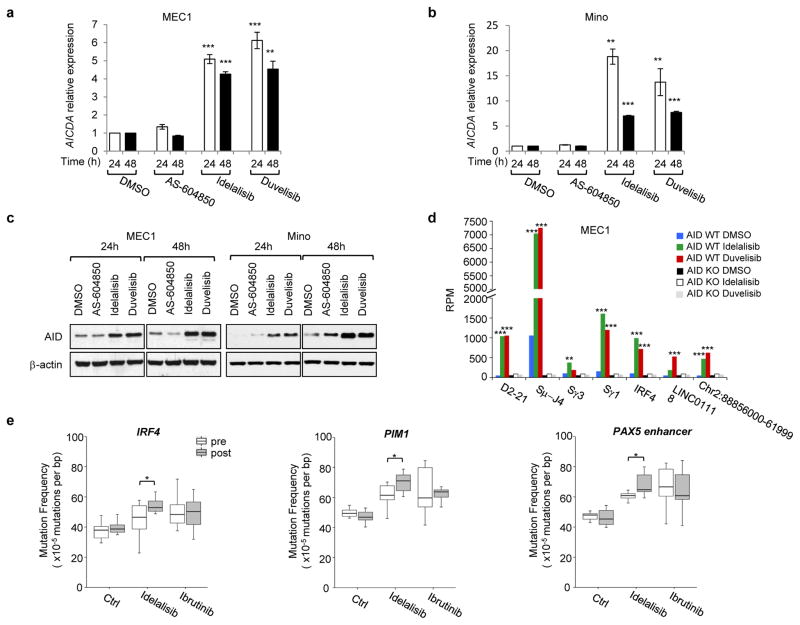
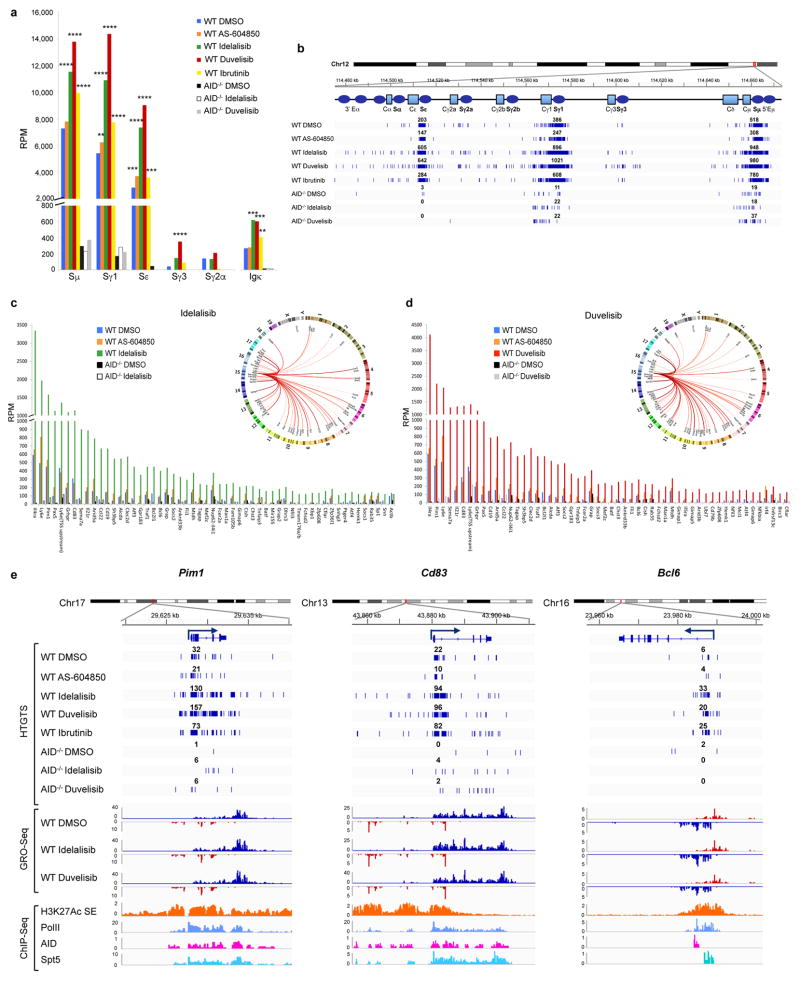
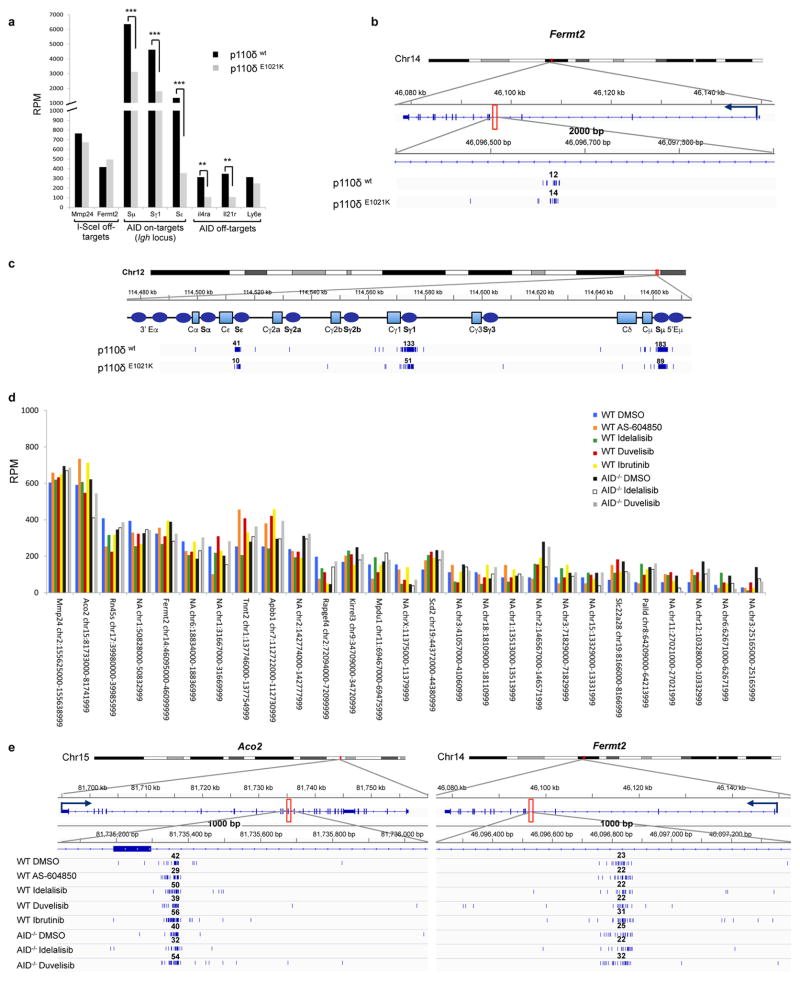
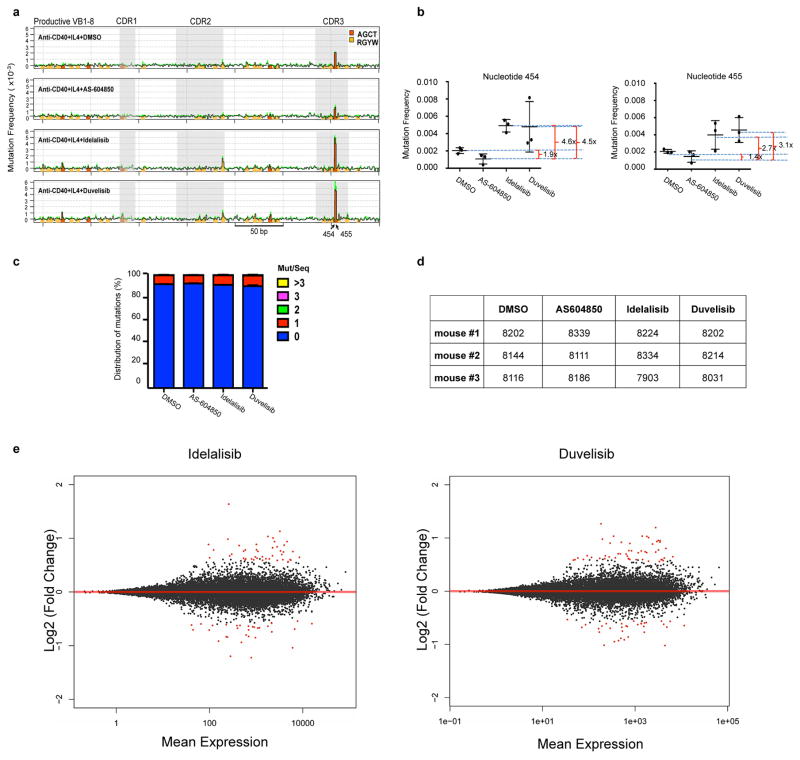
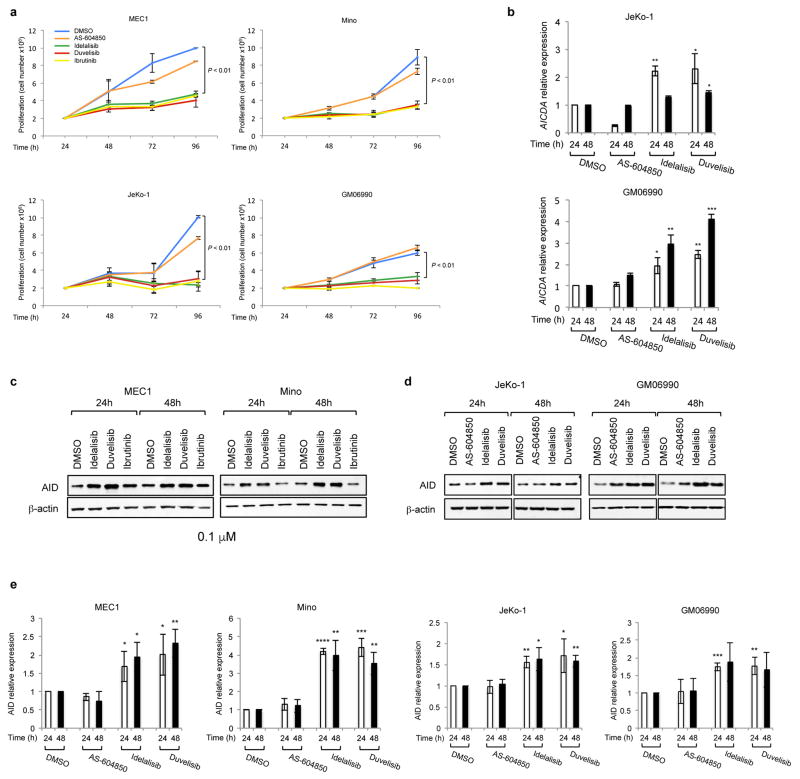
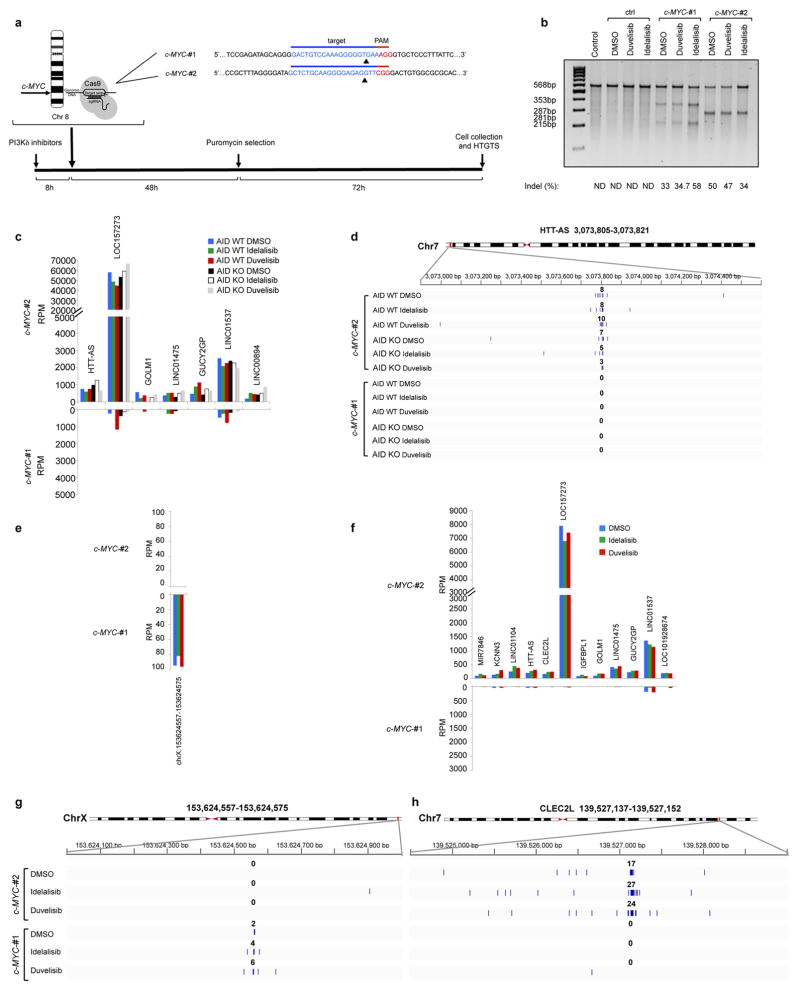
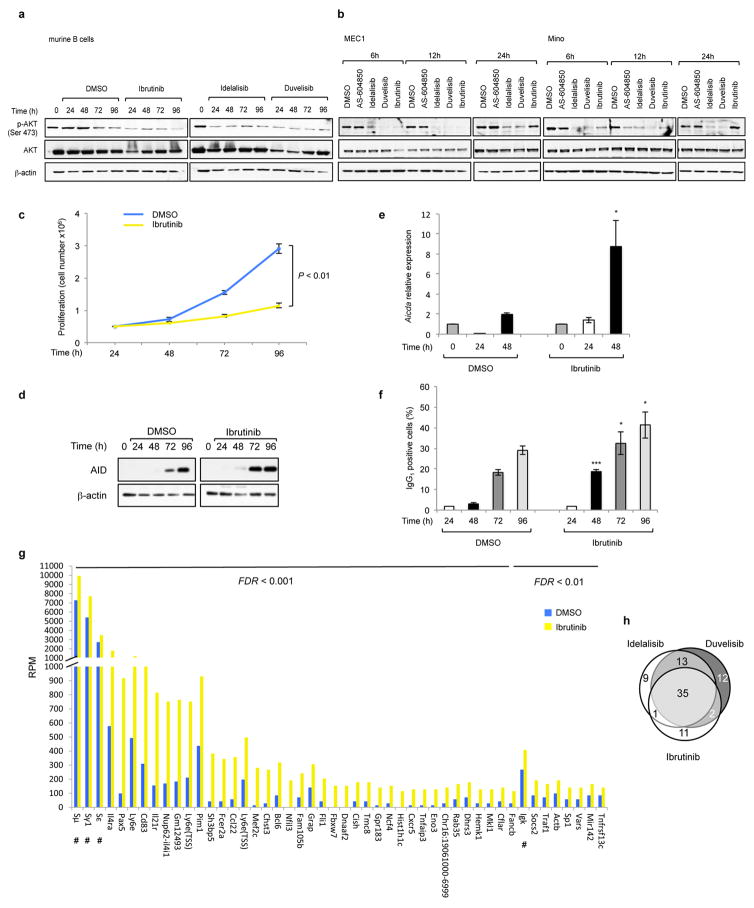
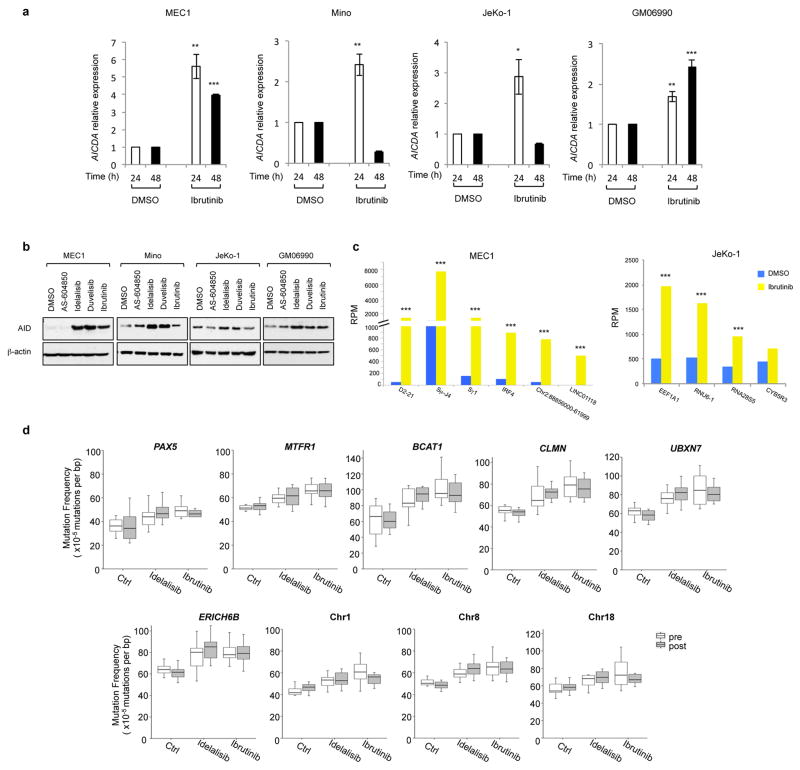
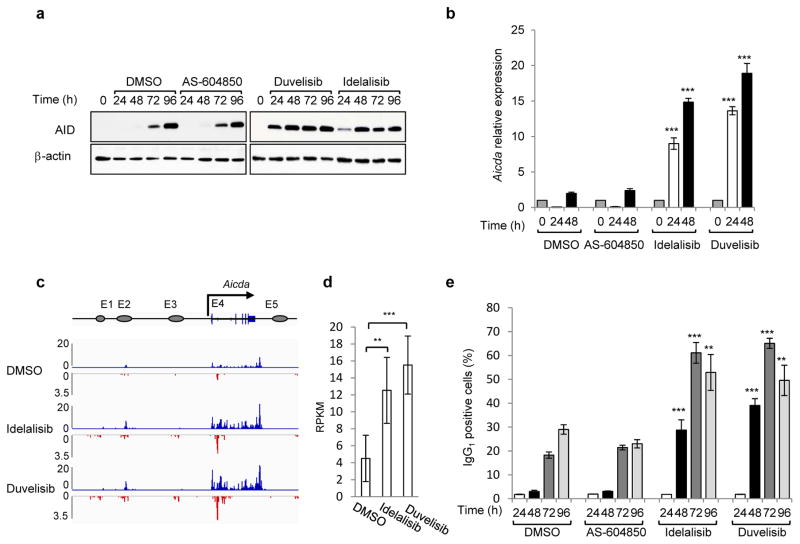
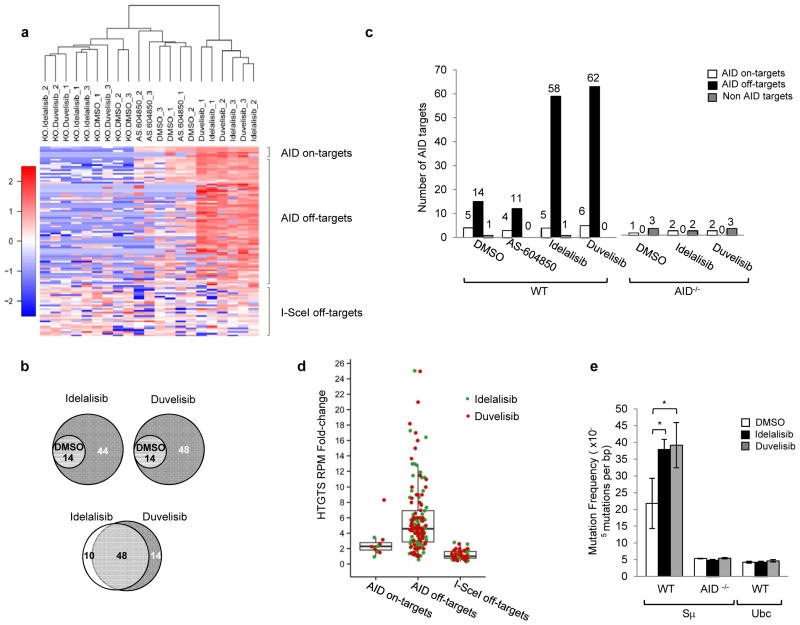
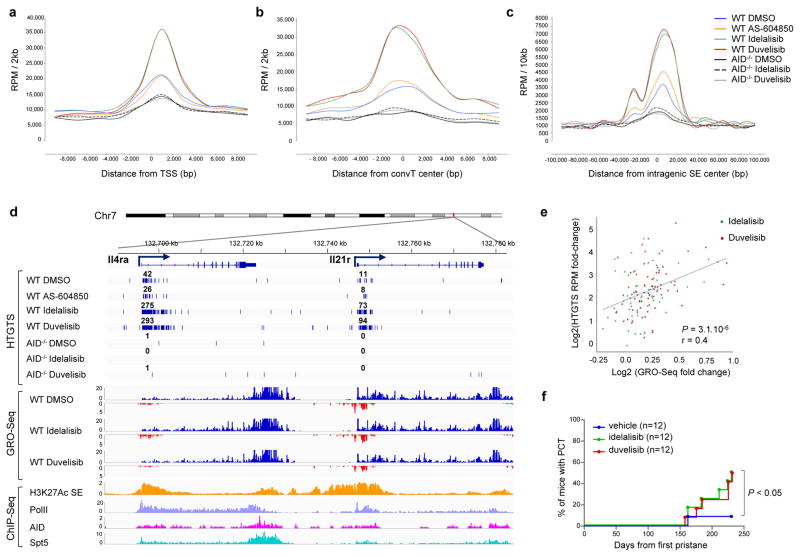
Activation-induced cytidine deaminase (AID) is a B-cell-specific enzyme that targets immunoglobulin genes to initiate class switch recombination and somatic hypermutation. In addition, through off-target activity, AID has a much broader effect on genomic instability by initiating oncogenic chromosomal translocations and mutations involved in the development and progression of lymphoma. AID expression is tightly regulated in B cells and its overexpression leads to enhanced genomic instability and lymphoma formation. The phosphatidylinositol 3-kinase δ (PI3Kδ) pathway regulates AID by suppressing its expression in B cells. Drugs for leukaemia or lymphoma therapy such as idelalisib, duvelisib and ibrutinib block PI3Kδ activity directly or indirectly, potentially affecting AID expression and, consequently, genomic stability in B cells. Here we show that treatment of primary mouse B cells with idelalisib or duvelisib, and to a lesser extent ibrutinib, enhanced the expression of AID and increased somatic hypermutation and chromosomal translocation frequency to the Igh locus and to several AID off-target sites. Both of these effects were completely abrogated in AID-deficient B cells. PI3Kδ inhibitors or ibrutinib increased the formation of AID-dependent tumours in pristane-treated mice. Consistently, PI3Kδ inhibitors enhanced AID expression and translocation frequency to IGH and AID off-target sites in human chronic lymphocytic leukaemia and mantle cell lymphoma cell lines, and patients treated with idelalisib, but not ibrutinib, showed increased somatic hypermutation in AID off-targets. In summary, we show that PI3Kδ or Bruton's tyrosine kinase inhibitors increase genomic instability in normal and neoplastic B cells by an AID-dependent mechanism. This effect should be carefully considered, as such inhibitors can be administered to patients for years.
激活诱导的胞苷脱氨酶(AID)是一种B细胞特异性酶,它作用于免疫球蛋白基因以启动类别转换重组和体细胞高频突变。此外,通过脱靶活性,AID通过引发致癌染色体易位以及与淋巴瘤发生和发展相关的突变,对基因组不稳定性产生更广泛的影响。AID在B细胞中的表达受到严格调控,其过表达会导致基因组不稳定性增强和淋巴瘤形成。磷脂酰肌醇3-激酶δ(PI3Kδ)途径通过抑制AID在B细胞中的表达来对其进行调节。用于白血病或淋巴瘤治疗的药物,如idelalisib、duvelisib和ibrutinib,直接或间接阻断PI3Kδ活性,可能影响AID表达,进而影响B细胞中的基因组稳定性。在此,我们表明,用idelalisib或duvelisib处理原代小鼠B细胞,以及在较小程度上用ibrutinib处理,会增强AID的表达,并增加体细胞高频突变以及到Igh基因座和几个AID脱靶位点的染色体易位频率。这两种效应在AID缺陷的B细胞中完全消除。PI3Kδ抑制剂或ibrutinib会增加在用 pristane处理的小鼠中AID依赖性肿瘤的形成。同样,PI3Kδ抑制剂会增强AID在人慢性淋巴细胞白血病和套细胞淋巴瘤细胞系中向IGH和AID脱靶位点的表达和易位频率,接受idelalisib而非ibrutinib治疗的患者在AID脱靶位点显示体细胞高频突变增加。总之,我们表明PI3Kδ或布鲁顿酪氨酸激酶抑制剂通过AID依赖性机制增加正常和肿瘤性B细胞中的基因组不稳定性。由于此类抑制剂可能会给患者使用数年,因此应仔细考虑这种效应。