Duffy Craig W, Ba Hampate, Assefa Samuel, Ahouidi Ambroise D, Deh Yacine B, Tandia Abderahmane, Kirsebom Freja C M, Kwiatkowski Dominic P, Conway David J
Department of Pathogen Molecular Biology, London School of Hygiene & Tropical Medicine, Keppel St, London, WC1E 7HT, UK.
Institut National de Recherche en Sante Publique, Nouakchott, Mauritania.
Mol Ecol. 2017 Jun;26(11):2880-2894. doi: 10.1111/mec.14066. Epub 2017 Mar 15.
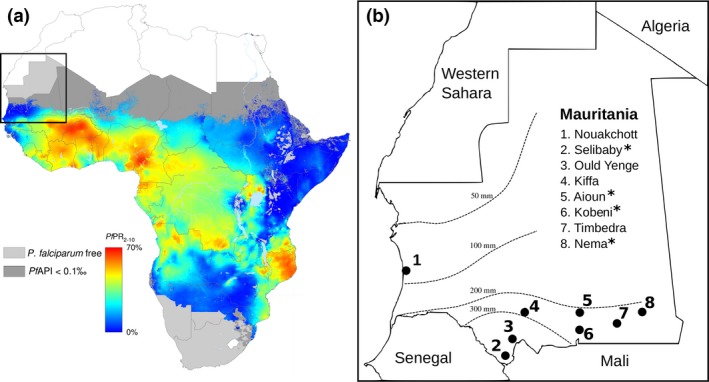
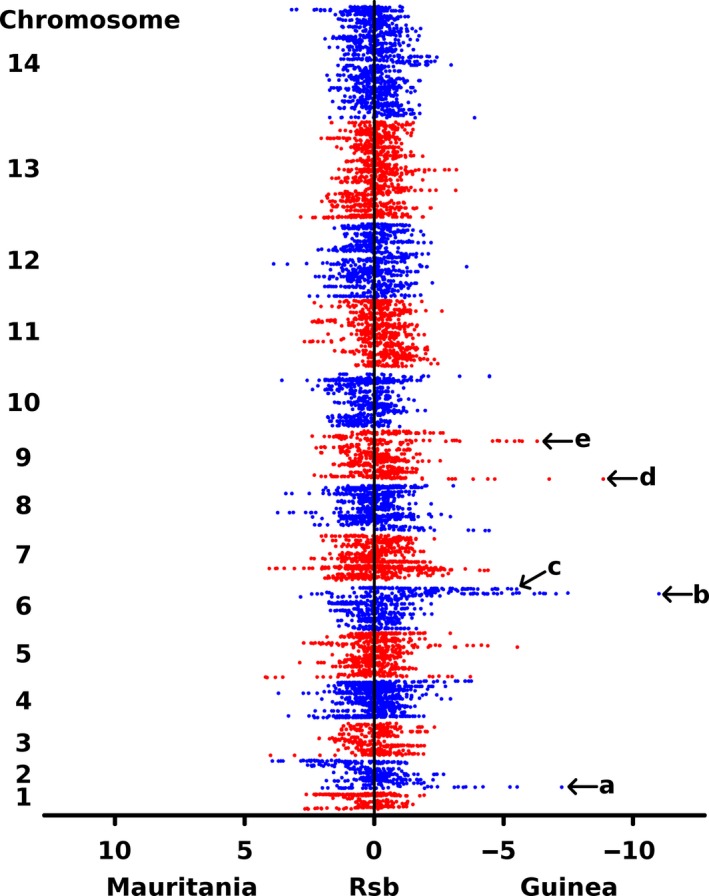
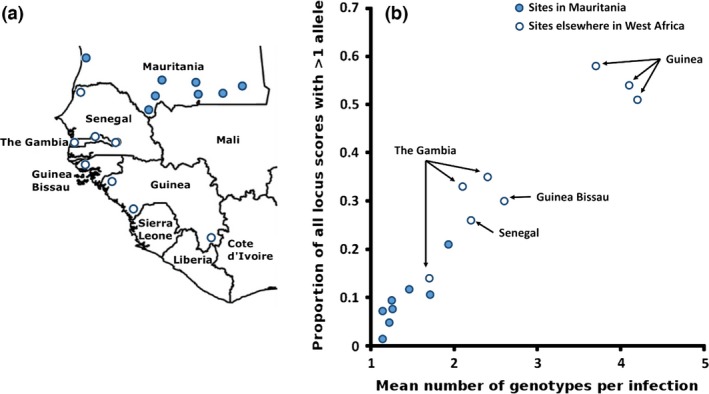
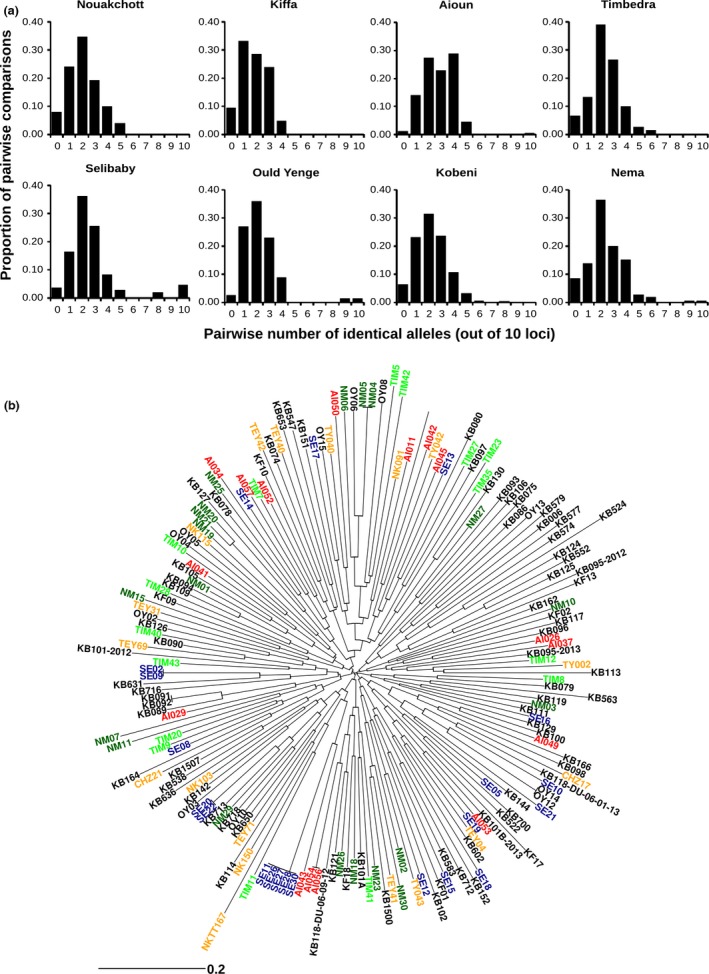
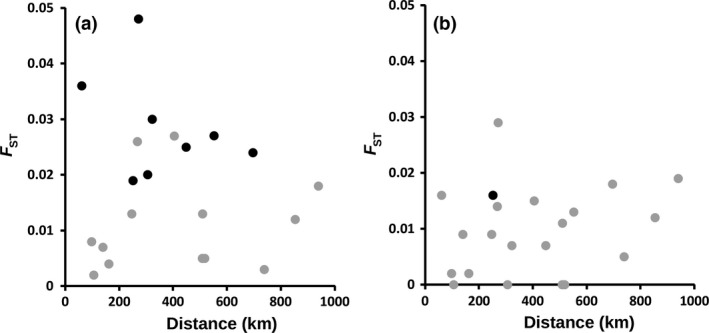
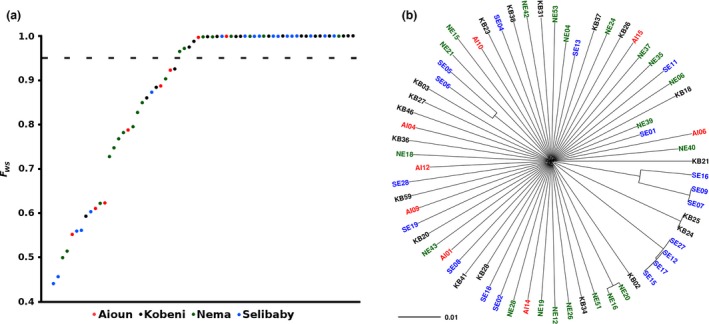
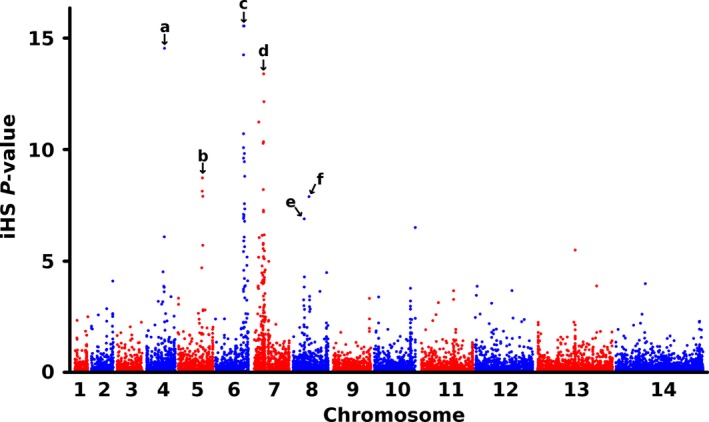
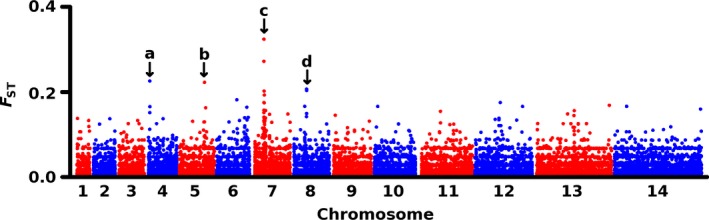
To determine whether the major human malaria parasite Plasmodium falciparum exhibits fragmented population structure or local adaptation at the northern limit of its African distribution where the dry Sahel zone meets the Sahara, samples were collected from diverse locations within Mauritania over a range of ~1000 km. Microsatellite genotypes were obtained for 203 clinical infection samples from eight locations, and Illumina paired-end sequences were obtained to yield high coverage genomewide single nucleotide polymorphism (SNP) data for 65 clinical infection samples from four locations. Most infections contained single parasite genotypes, reflecting low rates of transmission and superinfection locally, in contrast to the situation seen in population samples from countries further south. A minority of infections shared related or identical genotypes locally, indicating some repeated transmission of parasite clones without recombination. This caused some multilocus linkage disequilibrium and local divergence, but aside from the effect of repeated genotypes there was minimal differentiation between locations. Several chromosomal regions had elevated integrated haplotype scores (|iHS|) indicating recent selection, including those containing drug resistance genes. A genomewide F scan comparison with previous sequence data from an area in West Africa with higher infection endemicity indicates that regional gene flow prevents genetic isolation, but revealed allele frequency differentiation at three drug resistance loci and an erythrocyte invasion ligand gene. Contrast of extended haplotype signatures revealed none to be unique to Mauritania. Discrete foci of infection on the edge of the Sahara are genetically highly connected to the wider continental parasite population, and local elimination would be difficult to achieve without very substantial reduction in malaria throughout the region.
为了确定主要的人类疟原虫恶性疟原虫在其非洲分布的北部边界(干旱的萨赫勒地区与撒哈拉沙漠交汇之处)是否呈现出碎片化的种群结构或局部适应性,在毛里塔尼亚境内约1000公里范围内的不同地点采集了样本。从八个地点的203份临床感染样本中获得了微卫星基因型,并从四个地点的65份临床感染样本中获得了Illumina双端序列,以产生全基因组高覆盖率的单核苷酸多态性(SNP)数据。与更南部国家的种群样本情况相反,大多数感染包含单一寄生虫基因型,这反映出当地传播率和重复感染率较低。少数感染在当地共享相关或相同的基因型,表明寄生虫克隆在没有重组的情况下有一些重复传播。这导致了一些多位点连锁不平衡和局部差异,但除了重复基因型的影响外,各地点之间的分化极小。几个染色体区域的综合单倍型得分(|iHS|)升高,表明近期受到了选择,包括那些含有耐药基因的区域。与来自西非一个感染流行率较高地区的先前序列数据进行全基因组F扫描比较表明,区域基因流可防止基因隔离,但在三个耐药位点和一个红细胞入侵配体基因处发现了等位基因频率差异。扩展单倍型特征的对比显示,没有一个是毛里塔尼亚特有的。撒哈拉沙漠边缘的离散感染病灶在基因上与更广泛的大陆寄生虫种群高度相连,如果不大幅降低整个地区的疟疾发病率,很难实现局部消除。