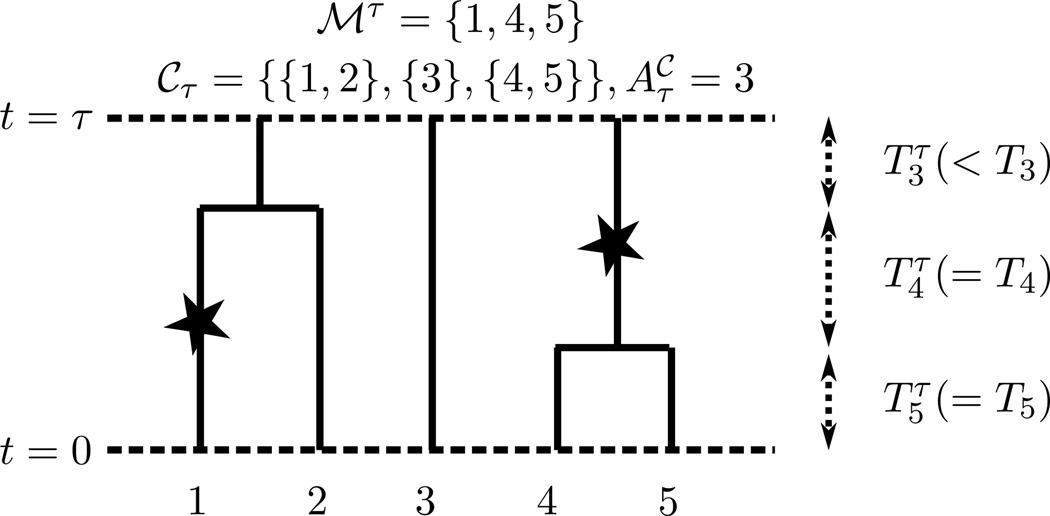
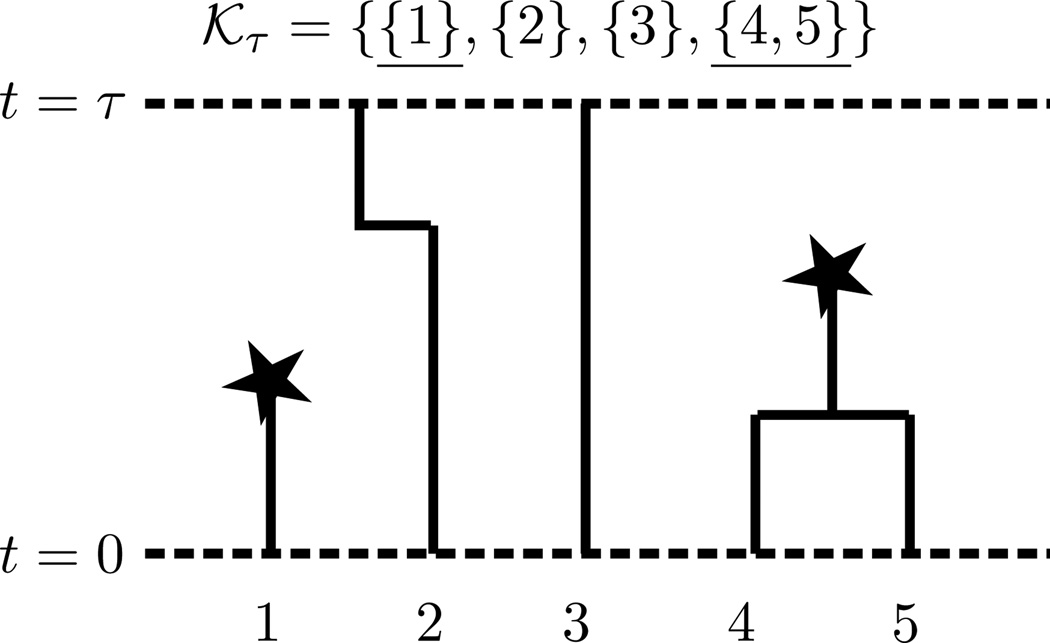
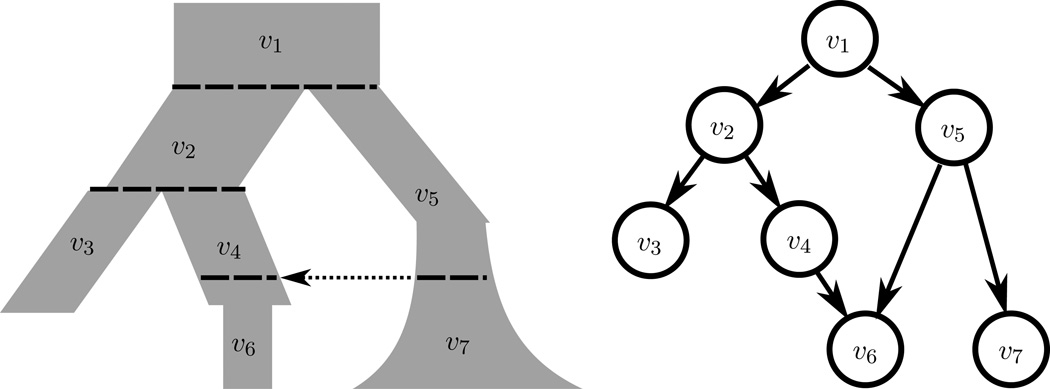
Kamm John A, Terhorst Jonathan, Song Yun S
Department of Statistics, University of California, Berkeley.
Departments of EECS, Statistics, and Integrative Biology, University of California, Berkeley.
J Comput Graph Stat. 2017;26(1):182-194. doi: 10.1080/10618600.2016.1159212. Epub 2017 Feb 16.
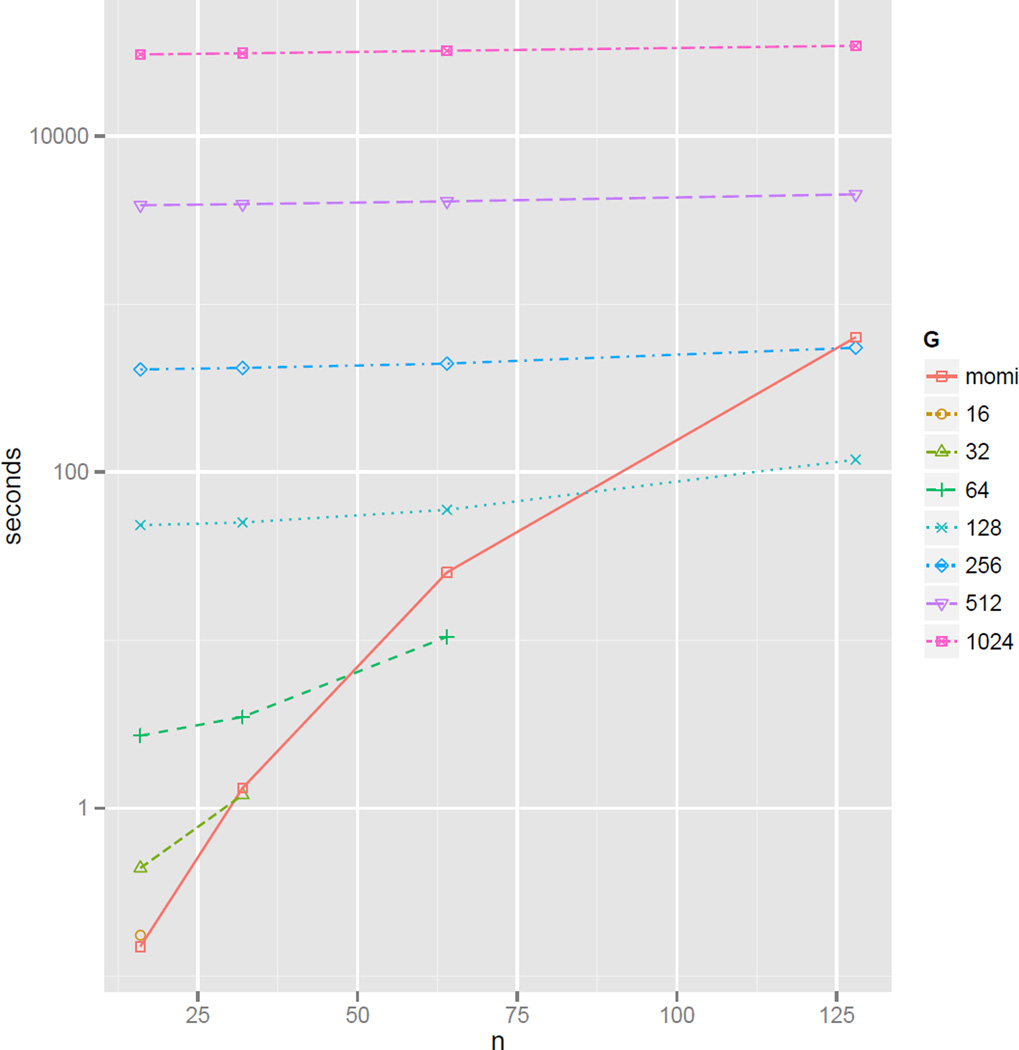
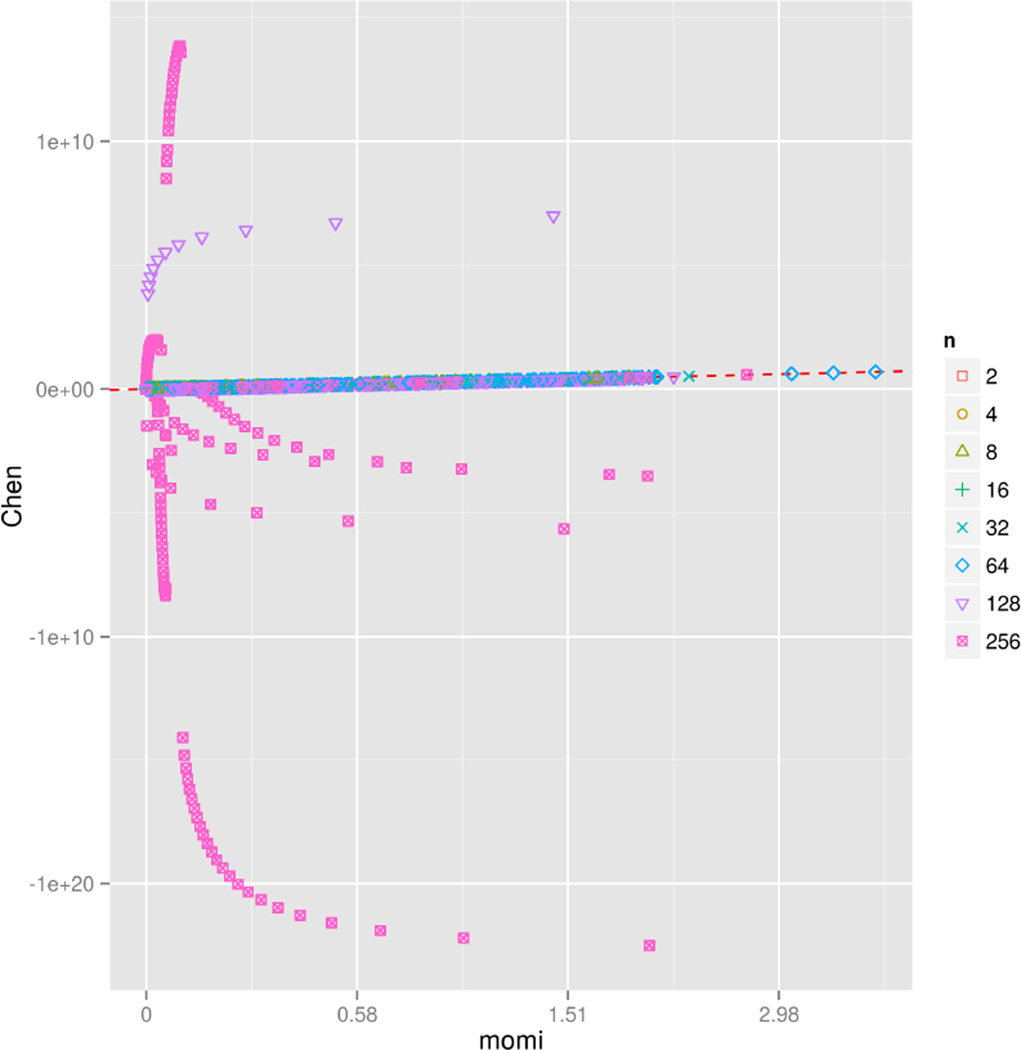
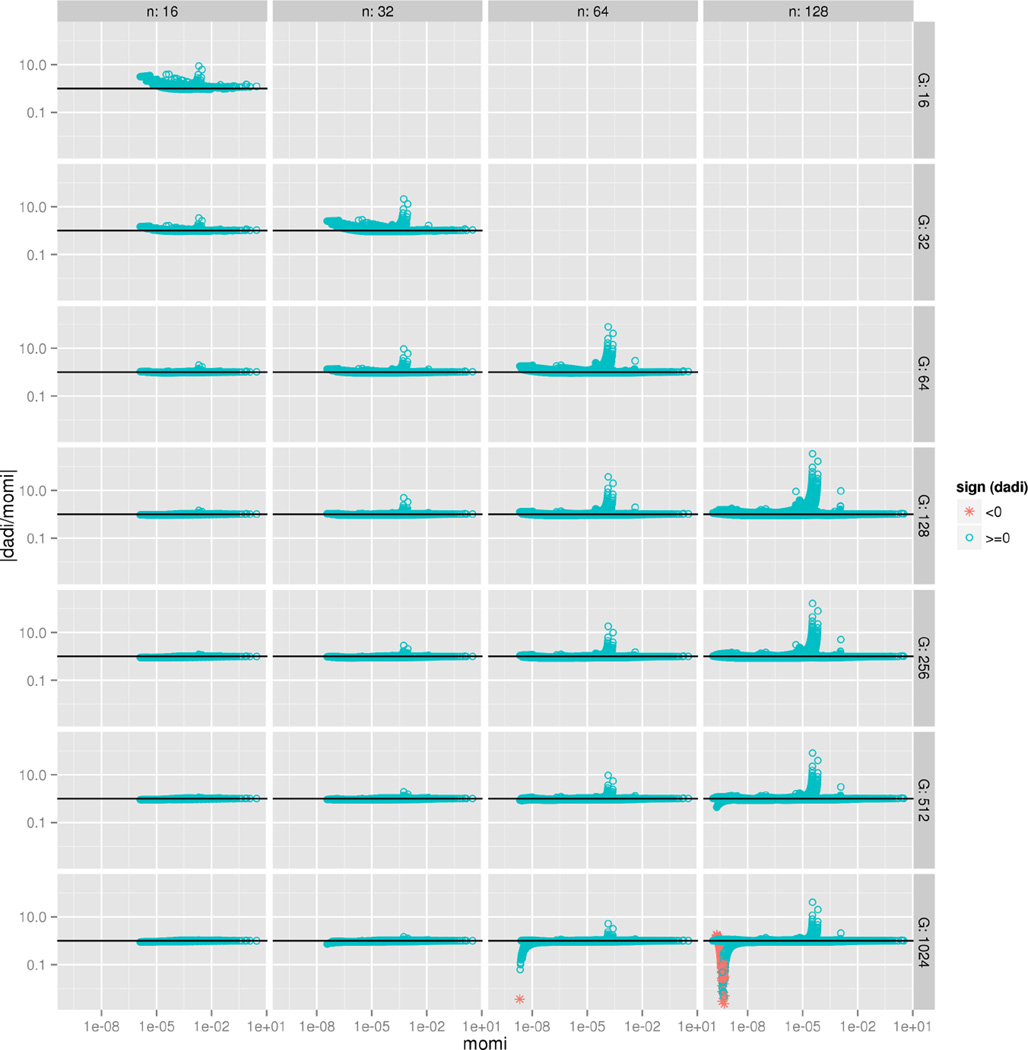
A wide range of studies in population genetics have employed the sample frequency spectrum (SFS), a summary statistic which describes the distribution of mutant alleles at a polymorphic site in a sample of DNA sequences and provides a highly efficient dimensional reduction of large-scale population genomic variation data. Recently, there has been much interest in analyzing the joint SFS data from multiple populations to infer parameters of complex demographic histories, including variable population sizes, population split times, migration rates, admixture proportions, and so on. SFS-based inference methods require accurate computation of the expected SFS under a given demographic model. Although much methodological progress has been made, existing methods suffer from numerical instability and high computational complexity when multiple populations are involved and the sample size is large. In this paper, we present new analytic formulas and algorithms that enable accurate, efficient computation of the expected joint SFS for thousands of individuals sampled from hundreds of populations related by a complex demographic model with arbitrary population size histories (including piecewise-exponential growth). Our results are implemented in a new software package called (MOran Models for Inference). Through an empirical study we demonstrate our improvements to numerical stability and computational complexity.
群体遗传学中的大量研究都采用了样本频率谱(SFS),它是一种汇总统计量,描述了DNA序列样本中多态性位点处突变等位基因的分布,并能对大规模群体基因组变异数据进行高效的降维处理。最近,人们对分析来自多个群体的联合SFS数据以推断复杂人口历史参数产生了浓厚兴趣,这些参数包括可变的群体大小、群体分裂时间、迁移率、混合比例等等。基于SFS的推断方法需要在给定的人口模型下准确计算预期的SFS。尽管在方法上已经取得了很大进展,但当涉及多个群体且样本量较大时,现有方法存在数值不稳定性和高计算复杂性的问题。在本文中,我们提出了新的解析公式和算法,能够对从数百个通过具有任意群体大小历史(包括分段指数增长)的复杂人口模型相关的群体中抽取的数千个个体准确、高效地计算预期的联合SFS。我们的结果在一个名为 (用于推断的莫兰模型)的新软件包中得以实现。通过实证研究,我们展示了在数值稳定性和计算复杂性方面的改进。