Huang Tao, Mi Hong, Lin Cheng-Yuan, Zhao Ling, Zhong Linda L D, Liu Feng-Bin, Zhang Ge, Lu Ai-Ping, Bian Zhao-Xiang
Lab of Brain and Gut Research, School of Chinese Medicine, Hong Kong Baptist University, 7 Baptist University Road, Hong Kong, People's Republic of China.
Department of Gastroenterology, the First Affiliated Hospital of Guangzhou University of Chinese Medicine, Guangzhou, 510405, People's Republic of China.
BMC Bioinformatics. 2017 Mar 11;18(1):165. doi: 10.1186/s12859-017-1586-z.
Many computational approaches have been used for target prediction, including machine learning, reverse docking, bioactivity spectra analysis, and chemical similarity searching. Recent studies have suggested that chemical similarity searching may be driven by the most-similar ligand. However, the extent of bioactivity of most-similar ligands has been oversimplified or even neglected in these studies, and this has impaired the prediction power.
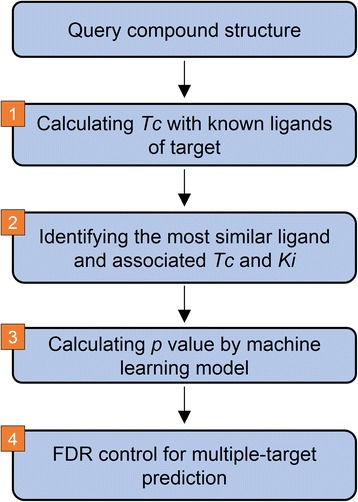
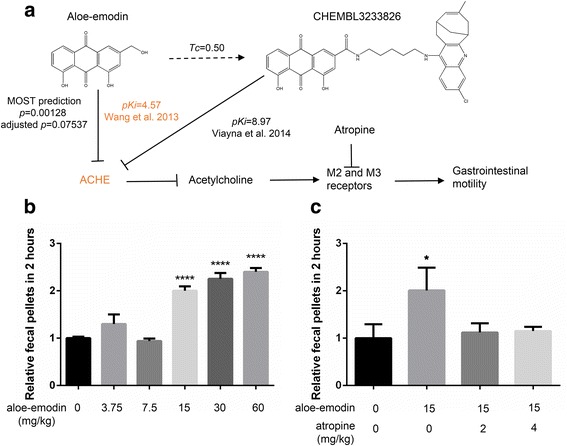
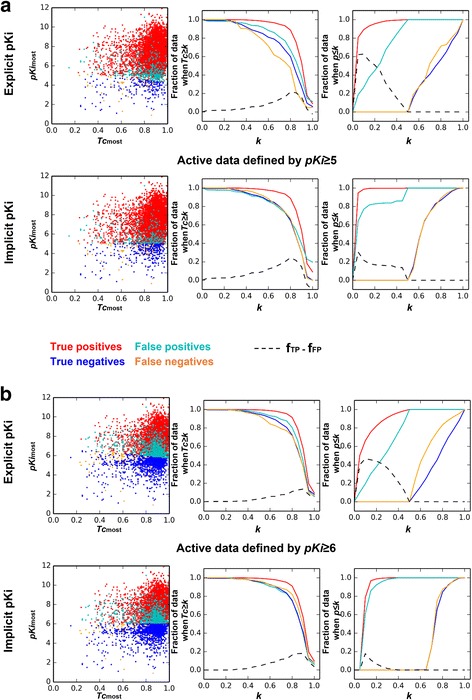
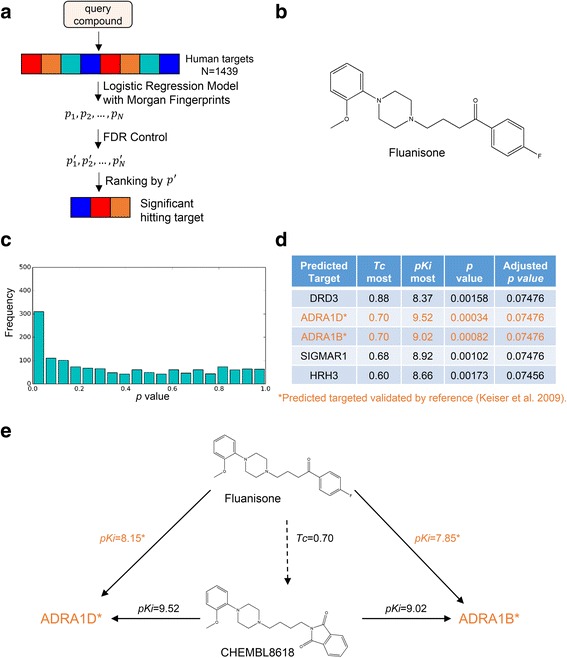
Here we propose the MOst-Similar ligand-based Target inference approach, namely MOST, which uses fingerprint similarity and explicit bioactivity of the most-similar ligands to predict targets of the query compound. Performance of MOST was evaluated by using combinations of different fingerprint schemes, machine learning methods, and bioactivity representations. In sevenfold cross-validation with a benchmark Ki dataset from CHEMBL release 19 containing 61,937 bioactivity data of 173 human targets, MOST achieved high average prediction accuracy (0.95 for pKi ≥ 5, and 0.87 for pKi ≥ 6). Morgan fingerprint was shown to be slightly better than FP2. Logistic Regression and Random Forest methods performed better than Naïve Bayes. In a temporal validation, the Ki dataset from CHEMBL19 were used to train models and predict the bioactivity of newly deposited ligands in CHEMBL20. MOST also performed well with high accuracy (0.90 for pKi ≥ 5, and 0.76 for pKi ≥ 6), when Logistic Regression and Morgan fingerprint were employed. Furthermore, the p values associated with explicit bioactivity were found be a robust index for removing false positive predictions. Implicit bioactivity did not offer this capability. Finally, p values generated with Logistic Regression, Morgan fingerprint and explicit activity were integrated with a false discovery rate (FDR) control procedure to reduce false positives in multiple-target prediction scenario, and the success of this strategy it was demonstrated with a case of fluanisone. In the case of aloe-emodin's laxative effect, MOST predicted that acetylcholinesterase was the mechanism-of-action target; in vivo studies validated this prediction.
Using the MOST approach can result in highly accurate and robust target prediction. Integrated with a FDR control procedure, MOST provides a reliable framework for multiple-target inference. It has prospective applications in drug repurposing and mechanism-of-action target prediction.
许多计算方法已被用于靶点预测,包括机器学习、反向对接、生物活性谱分析和化学相似性搜索。最近的研究表明,化学相似性搜索可能由最相似的配体驱动。然而,在这些研究中,最相似配体的生物活性程度被过度简化甚至被忽视,这削弱了预测能力。
在此,我们提出了基于最相似配体的靶点推断方法,即MOST,它利用指纹相似性和最相似配体的明确生物活性来预测查询化合物的靶点。通过使用不同指纹方案、机器学习方法和生物活性表示的组合来评估MOST的性能。在对来自CHEMBL版本19的包含173个人类靶点的61937个生物活性数据的基准Ki数据集进行七折交叉验证时,MOST实现了较高的平均预测准确率(对于pKi≥5为0.95,对于pKi≥6为0.87)。结果表明,摩根指纹略优于FP2。逻辑回归和随机森林方法的表现优于朴素贝叶斯。在一次时间验证中,使用CHEMBL19的Ki数据集训练模型并预测CHEMBL20中新存入配体的生物活性。当采用逻辑回归和摩根指纹时,MOST也表现良好,准确率较高(对于pKi≥5为0.90,对于pKi≥6为0.76)。此外,发现与明确生物活性相关的p值是去除假阳性预测的可靠指标。隐式生物活性不具备此能力。最后,将通过逻辑回归、摩根指纹和明确活性生成的p值与错误发现率(FDR)控制程序相结合,以减少多靶点预测场景中的假阳性,并且以氟胺酮为例证明了该策略的成功。在芦荟大黄素的通便作用案例中,MOST预测乙酰胆碱酯酶是其作用机制靶点;体内研究验证了这一预测。
使用MOST方法可实现高度准确和稳健的靶点预测。与FDR控制程序相结合,MOST为多靶点推断提供了一个可靠的框架。它在药物再利用和作用机制靶点预测方面具有潜在应用。