MRC SGDP Centre, Institute of Psychiatry, Psychology &Neuroscience, King's College London, London, SE5 8AF, UK.
Sci Rep. 2017 Mar 13;7:38837. doi: 10.1038/srep38837.
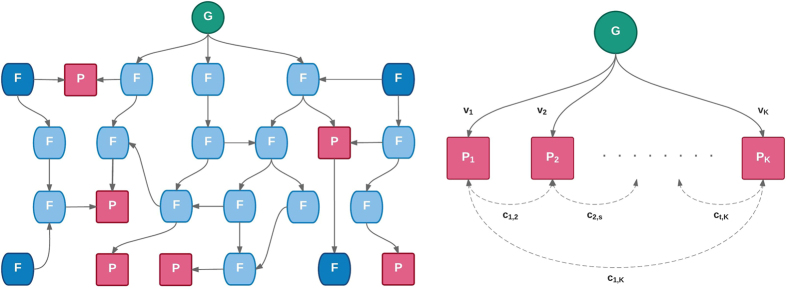
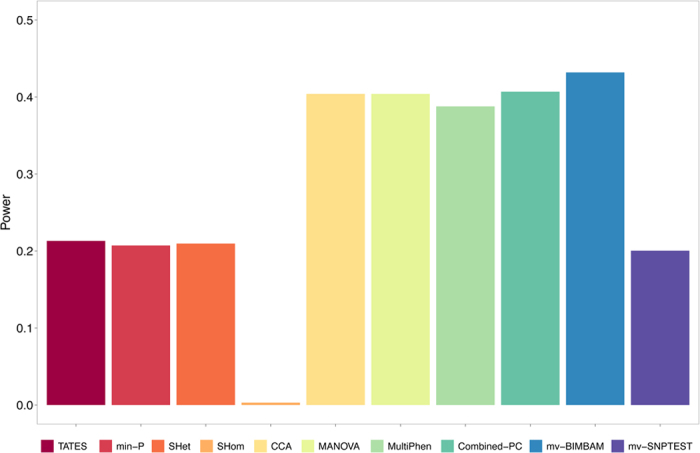
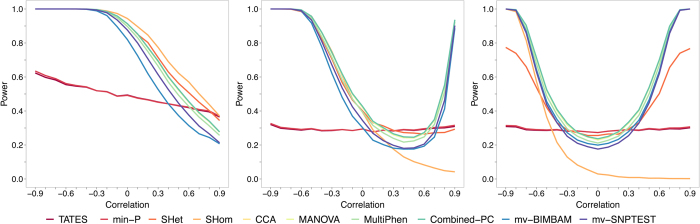
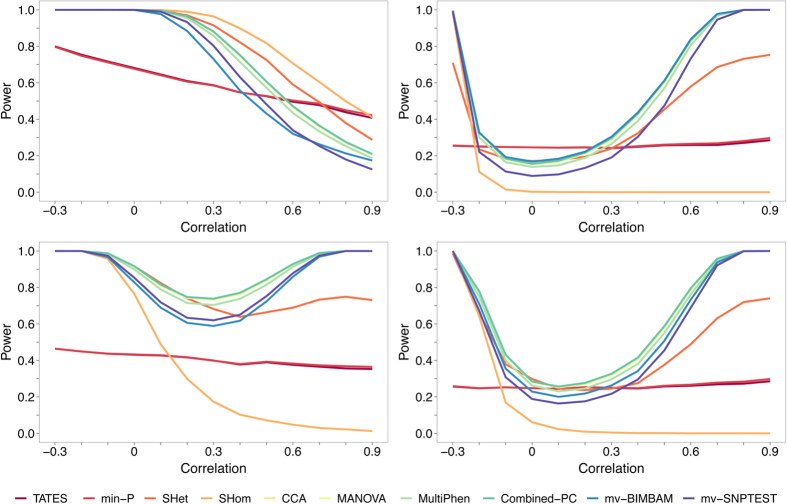
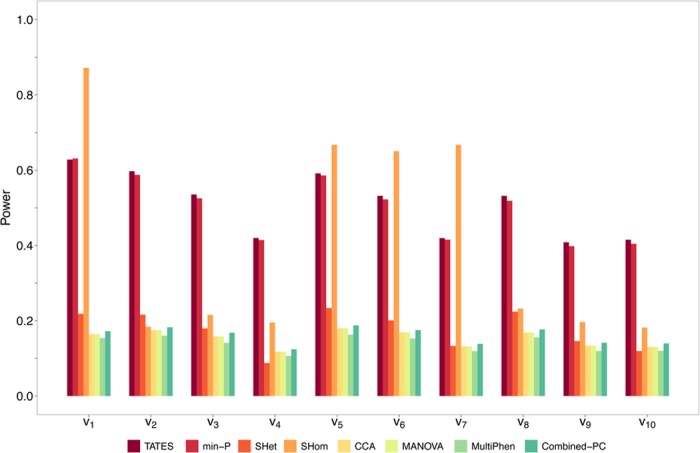
Burgeoning availability of genome-wide association study (GWAS) results and national biobank data has led to growing interest in performing multi-trait genetic analyses. Numerous multi-trait GWAS methods that exploit either summary statistics or individual-level data have been developed, but their relative performance is unclear. Here we develop a simulation framework to model the complex networks underlying multivariate genetic epidemiology, enabling the vast model space of genetic effects on multiple correlated traits to be explored systematically. We perform a comprehensive comparison of the leading multi-trait GWAS methods, finding: (1) method performance is highly sensitive to the specific combination of genetic effects and phenotypic correlations, (2) most of the current multivariate methods have remarkably similar statistical power, and (3) multivariate methods may offer a substantial increase in the discovery of genetic variants over the standard univariate approach. We believe our findings offer the clearest picture to date of the relative performance of multi-trait GWAS methods and act as a guide for method selection. We provide a web application and open-source software program implementing our simulation framework, for: (i) further benchmarking of multivariate GWAS methods, (ii) power calculations for multivariate genetic studies, and (iii) generating data for testing any multivariate method in genetic epidemiology.
全基因组关联研究(GWAS)结果和全国生物库数据的日益丰富,使得人们对进行多性状遗传分析的兴趣日益浓厚。已经开发了许多利用汇总统计数据或个体水平数据的多性状 GWAS 方法,但它们的相对性能尚不清楚。在这里,我们开发了一个模拟框架来模拟多变量遗传流行病学的复杂网络,从而能够系统地探索遗传效应对多个相关性状的巨大模型空间。我们对领先的多性状 GWAS 方法进行了全面比较,发现:(1)方法性能对遗传效应和表型相关性的具体组合高度敏感;(2)目前大多数多元方法具有非常相似的统计功效;(3)与标准单变量方法相比,多元方法可能会大大增加遗传变异的发现。我们相信,我们的研究结果提供了迄今为止对多性状 GWAS 方法相对性能的最清晰描述,并为方法选择提供了指导。我们提供了一个网络应用程序和开源软件程序来实现我们的模拟框架,用于:(i)进一步基准测试多元 GWAS 方法;(ii)多变量遗传研究的功效计算;(iii)生成用于测试遗传流行病学中任何多元方法的数据。