Department of Statistics, University of Wisconsin-Madison, Madison, Wisconsin, 53706, USA.
Biostatistics and Research Decision Sciences, Merck & Co., Inc., Rahway, New Jersey, 07065, USA.
Nat Commun. 2020 Jun 5;11(1):2850. doi: 10.1038/s41467-020-16591-0.
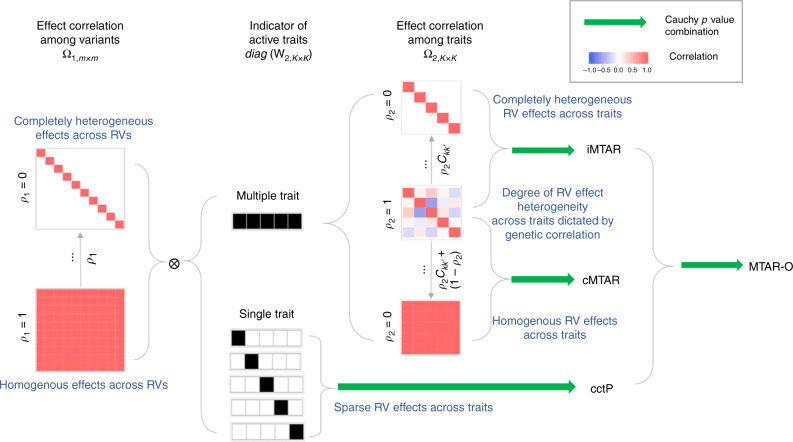
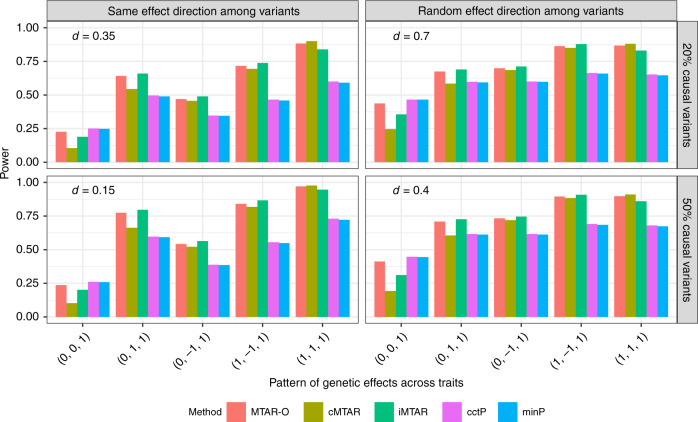
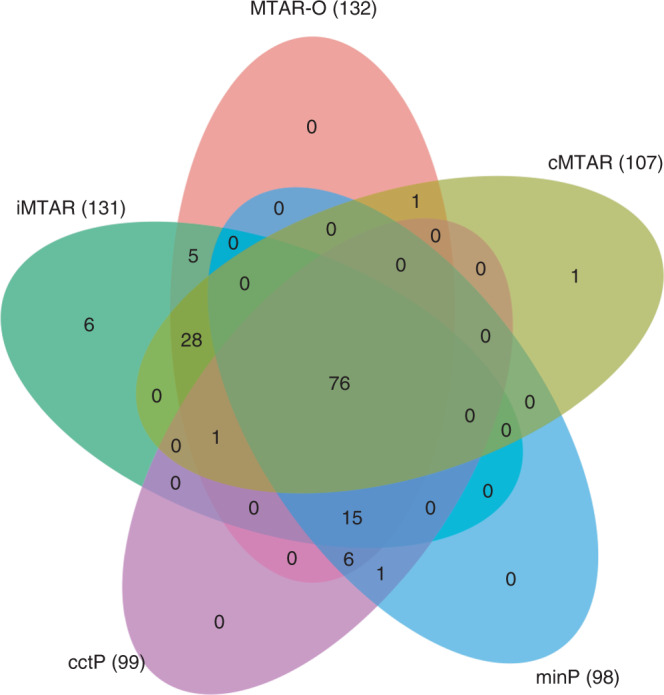
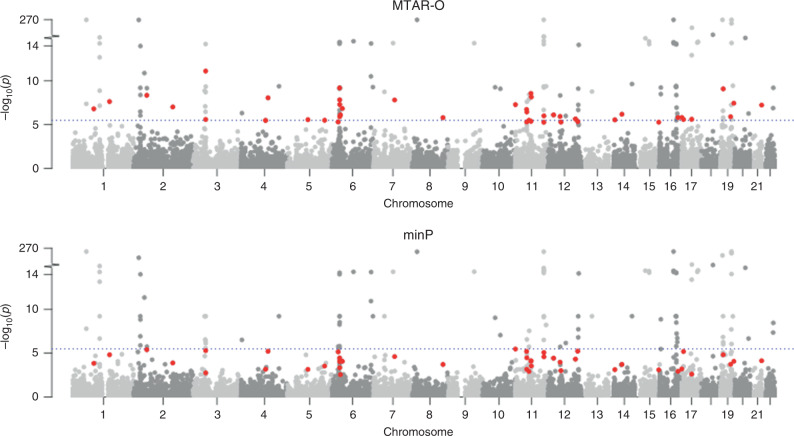
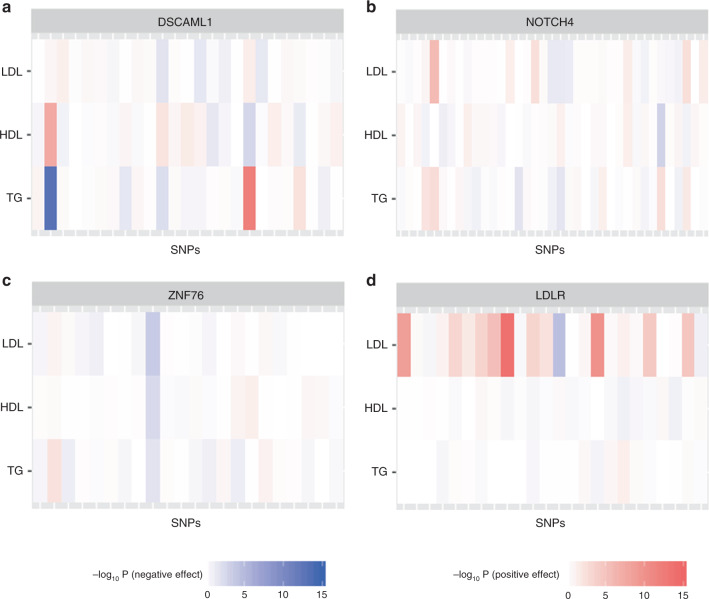
Integrating association evidence across multiple traits can improve the power of gene discovery and reveal pleiotropy. Most multi-trait analysis methods focus on individual common variants in genome-wide association studies. Here, we introduce multi-trait analysis of rare-variant associations (MTAR), a framework for joint analysis of association summary statistics between multiple rare variants and different traits. MTAR achieves substantial power gain by leveraging the genome-wide genetic correlation measure to inform the degree of gene-level effect heterogeneity across traits. We apply MTAR to rare-variant summary statistics for three lipid traits in the Global Lipids Genetics Consortium. 99 genome-wide significant genes were identified in the single-trait-based tests, and MTAR increases this to 139. Among the 11 novel lipid-associated genes discovered by MTAR, 7 are replicated in an independent UK Biobank GWAS analysis. Our study demonstrates that MTAR is substantially more powerful than single-trait-based tests and highlights the value of MTAR for novel gene discovery.
整合多个性状的关联证据可以提高基因发现的能力,并揭示多效性。大多数多性状分析方法都集中在全基因组关联研究中的个体常见变体上。在这里,我们引入了罕见变异关联的多性状分析(MTAR),这是一种联合分析多个罕见变异与不同性状之间关联汇总统计数据的框架。MTAR 通过利用全基因组遗传相关性度量来告知跨性状的基因水平效应异质性程度,从而获得实质性的功效增益。我们将 MTAR 应用于全球脂质遗传学联盟中三个脂质性状的罕见变异汇总统计数据。在基于单性状的检验中,确定了 99 个全基因组显著基因,而 MTAR 将其增加到 139 个。在 MTAR 发现的 11 个新的脂质相关基因中,有 7 个在独立的英国生物库 GWAS 分析中得到了复制。我们的研究表明,MTAR 比基于单性状的检验具有实质性的更高功效,并强调了 MTAR 对新基因发现的价值。