Muñoz Marina, Ríos-Chaparro Dora Inés, Patarroyo Manuel Alfonso, Ramírez Juan David
Grupo de Investigaciones Microbiológicas-UR (GIMUR), Programa de Biología, Facultad de Ciencias Naturales y Matemáticas, Universidad del Rosario, Carrera 24 # 63C - 69, Bogotá, Colombia.
Posgrado Interfacultades Doctorado en Biotecnología, Facultad de Ciencias, Universidad Nacional de Colombia, Bogotá, Colombia.
BMC Microbiol. 2017 Mar 14;17(1):62. doi: 10.1186/s12866-017-0969-7.
Multilocus sequence typing (MLST) is a highly discriminatory typing strategy; it is reproducible and scalable. There is a MLST scheme for Clostridium difficile (CD), a gram positive bacillus causing different pathologies of the gastrointestinal tract. This work was aimed at describing the frequency of sequence types (STs) and Clades (C) reported and evalute the intra-taxa diversity in the CD MLST database (CD-MLST-db) using an MLSA approach.
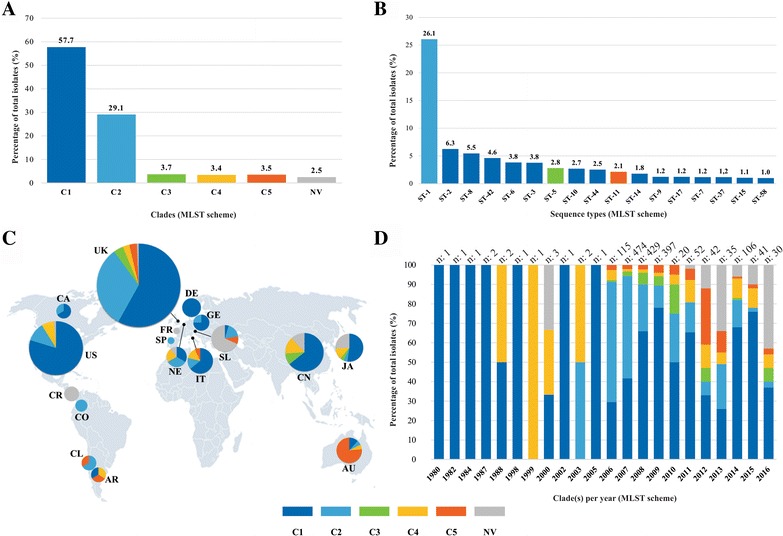
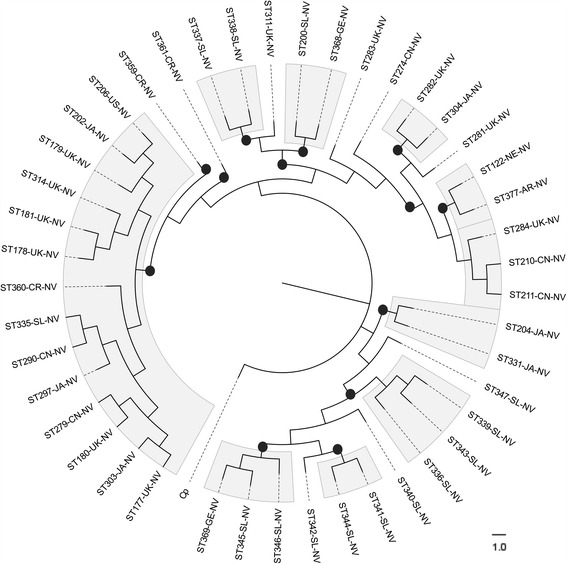
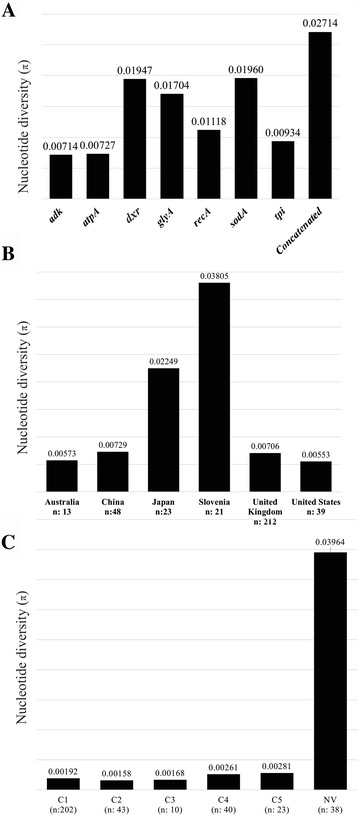
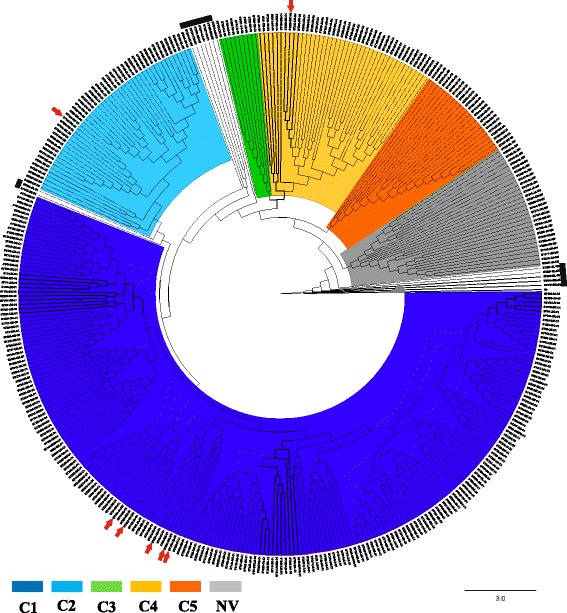
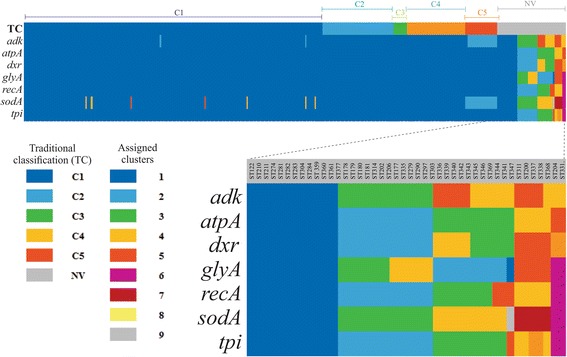
Analysis of 1778 available isolates showed that clade 1 (C1) was the most frequent worldwide (57.7%), followed by C2 (29.1%). Regarding sequence types (STs), it was found that ST-1, belonging to C2, was the most frequent. The isolates analysed came from 17 countries, mostly from the United Kingdom (UK) (1541 STs, 87.0%). The diversity of the seven housekeeping genes in the MLST scheme was evaluated, and alleles from the profiles (STs), for identifying CD population structure. It was found that adk and atpA are conserved genes allowing a limited amount of clusters to be discriminated; however, different genes such as drx, glyA and particularly sodA showed high diversity indexes and grouped CD populations in many clusters, suggesting that these genes' contribution to CD typing should be revised. It was identified that CD STs reported to date have a mostly clonal population structure with foreseen events of recombination; however, one group of STs was not assigned to a clade being highly different containing at least nine well-supported clusters, suggesting a greater amount of clades for CD.
This study shows the usefulness of CD-MLST-db as a tool for studying CD distribution and population structure, identifying the need for reviewing the usefulness of sodA as housekeeping gene within the MLST scheme and suggesting the existence of a greater amount of CD clades. The study also shows the plausible exchange of genetic material between STs, contributing towards intra-taxa genetic diversity.
多位点序列分型(MLST)是一种具有高度鉴别力的分型策略;它具有可重复性和可扩展性。对于艰难梭菌(CD)有一种MLST方案,艰难梭菌是一种革兰氏阳性杆菌,可引起胃肠道的不同病变。这项工作旨在描述已报告的序列类型(STs)和进化枝(C)的频率,并使用MLSA方法评估CD MLST数据库(CD - MLST - db)中的分类群内多样性。
对1778株可用分离株的分析表明,进化枝1(C1)在全球最为常见(57.7%),其次是C2(29.1%)。关于序列类型(STs),发现属于C2的ST - 1最为常见。分析的分离株来自17个国家,大部分来自英国(UK)(1541个STs,87.0%)。评估了MLST方案中七个管家基因的多样性,以及来自谱型(STs)的等位基因,以识别CD群体结构。发现adk和atpA是保守基因,只能区分有限数量的簇;然而,不同的基因如drx、glyA,特别是sodA显示出高多样性指数,并将CD群体分为许多簇,这表明这些基因对CD分型的贡献应重新评估。已确定,迄今为止报告的CD STs大多具有克隆群体结构,并伴有预期的重组事件;然而,一组STs未被分配到进化枝,它们高度不同,包含至少九个得到充分支持的簇,这表明CD可能存在更多的进化枝。
本研究表明CD - MLST - db作为研究CD分布和群体结构的工具是有用的,确定了需要重新审视sodA作为MLST方案中管家基因的有用性,并表明存在更多的CD进化枝。该研究还表明STs之间可能存在遗传物质交换,这有助于分类群内的遗传多样性。