Zhang J, Feng J-Y, Ni Y-L, Wen Y-J, Niu Y, Tamba C L, Yue C, Song Q, Zhang Y-M
State Key Laboratory of Crop Genetics and Germplasm Enhancement, Nanjing Agricultural University, Nanjing, China.
Soybean Genomics and Improvement Laboratory, Agricultural Research Service, United States Department of Agriculture, Beltsville, MD, USA.
Heredity (Edinb). 2017 Jun;118(6):517-524. doi: 10.1038/hdy.2017.8. Epub 2017 Mar 15.
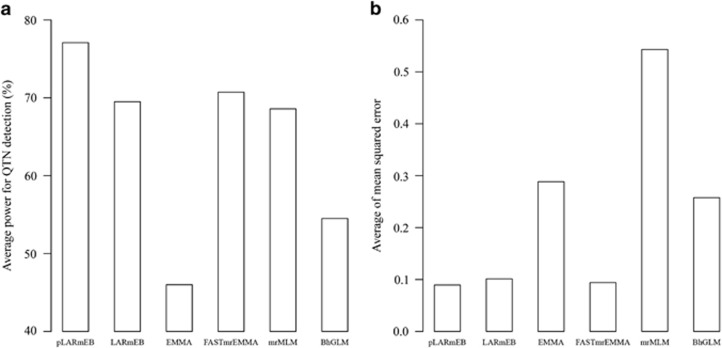
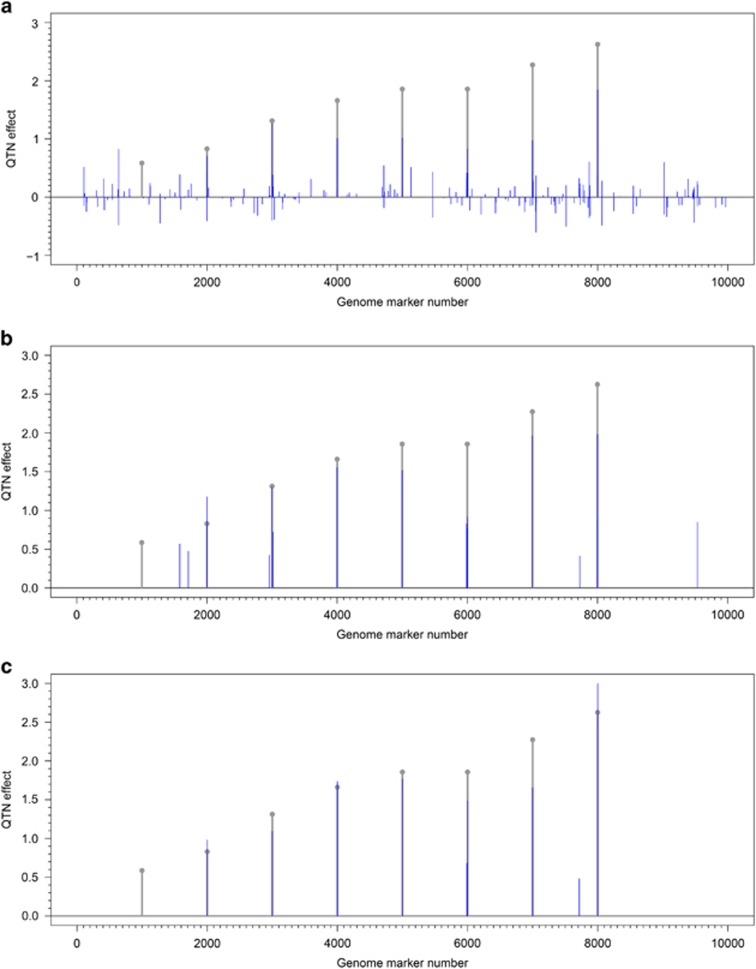
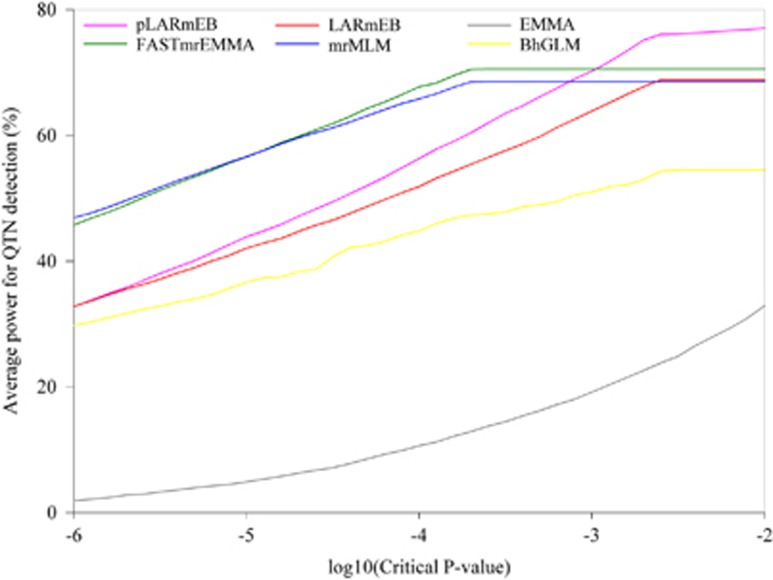
Multilocus genome-wide association studies (GWAS) have become the state-of-the-art procedure to identify quantitative trait nucleotides (QTNs) associated with complex traits. However, implementation of multilocus model in GWAS is still difficult. In this study, we integrated least angle regression with empirical Bayes to perform multilocus GWAS under polygenic background control. We used an algorithm of model transformation that whitened the covariance matrix of the polygenic matrix K and environmental noise. Markers on one chromosome were included simultaneously in a multilocus model and least angle regression was used to select the most potentially associated single-nucleotide polymorphisms (SNPs), whereas the markers on the other chromosomes were used to calculate kinship matrix as polygenic background control. The selected SNPs in multilocus model were further detected for their association with the trait by empirical Bayes and likelihood ratio test. We herein refer to this method as the pLARmEB (polygenic-background-control-based least angle regression plus empirical Bayes). Results from simulation studies showed that pLARmEB was more powerful in QTN detection and more accurate in QTN effect estimation, had less false positive rate and required less computing time than Bayesian hierarchical generalized linear model, efficient mixed model association (EMMA) and least angle regression plus empirical Bayes. pLARmEB, multilocus random-SNP-effect mixed linear model and fast multilocus random-SNP-effect EMMA methods had almost equal power of QTN detection in simulation experiments. However, only pLARmEB identified 48 previously reported genes for 7 flowering time-related traits in Arabidopsis thaliana.
多位点全基因组关联研究(GWAS)已成为识别与复杂性状相关的数量性状核苷酸(QTN)的最先进方法。然而,在GWAS中实施多位点模型仍然困难。在本研究中,我们将最小角回归与经验贝叶斯相结合,在多基因背景控制下进行多位点GWAS。我们使用了一种模型转换算法,该算法对多基因矩阵K和环境噪声的协方差矩阵进行了白化处理。一条染色体上的标记同时包含在一个多位点模型中,使用最小角回归来选择最有可能相关的单核苷酸多态性(SNP),而其他染色体上的标记用于计算亲缘关系矩阵作为多基因背景控制。通过经验贝叶斯和似然比检验进一步检测多位点模型中选定的SNP与性状的关联。我们在此将此方法称为pLARmEB(基于多基因背景控制的最小角回归加经验贝叶斯)。模拟研究结果表明,与贝叶斯分层广义线性模型、高效混合模型关联(EMMA)和最小角回归加经验贝叶斯相比,pLARmEB在QTN检测中更具功效,在QTN效应估计中更准确,假阳性率更低,所需计算时间更少。在模拟实验中,pLARmEB、多位点随机SNP效应混合线性模型和快速多位点随机SNP效应EMMA方法在QTN检测方面的功效几乎相同。然而,只有pLARmEB在拟南芥中鉴定出了48个先前报道的与7个开花时间相关性状的基因。