Kane Megan S, Davids Mariska, Bond Michelle R, Adams Christopher J, Grout Megan E, Phelps Ian G, O'Day Diana R, Dempsey Jennifer C, Li Xeuli, Golas Gretchen, Vezina Gilbert, Gunay-Aygun Meral, Hanover John A, Doherty Dan, He Miao, Malicdan May Christine V, Gahl William A, Boerkoel Cornelius F
NIH Undiagnosed Disease Program, Common Fund, Office of the Director, and National Human Genome Research Institute, National Institutes of Health, Bethesda, MD USA.
Inova Translational Medicine Institute, Inova Health System, Falls Church, VA USA.
Cilia. 2017 Mar 23;6:2. doi: 10.1186/s13630-017-0048-6. eCollection 2017.
The discovery of disease pathogenesis requires systematic agnostic screening of multiple homeostatic processes that may become deregulated. We illustrate this principle in the evaluation and diagnosis of a 5-year-old boy with Joubert syndrome type 10 (JBTS10). He carried the OFD1 mutation p.Gln886Lysfs*2 (NM_003611.2: c.2656del) and manifested features of Joubert syndrome.
We integrated exome sequencing, MALDI-TOF mass spectrometry analyses of plasma and cultured dermal fibroblasts glycomes, and full clinical evaluation of the proband. Analyses of cilia formation and lectin staining were performed by immunofluorescence. Measurement of cellular nucleotide sugar levels was performed with high-performance anion-exchange chromatography with pulsed amperometric detection. Statistical analyses utilized the Student's and Fisher's exact tests.
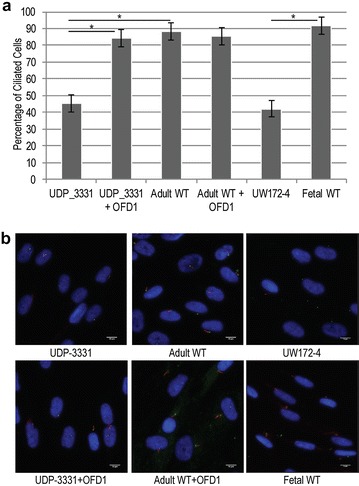
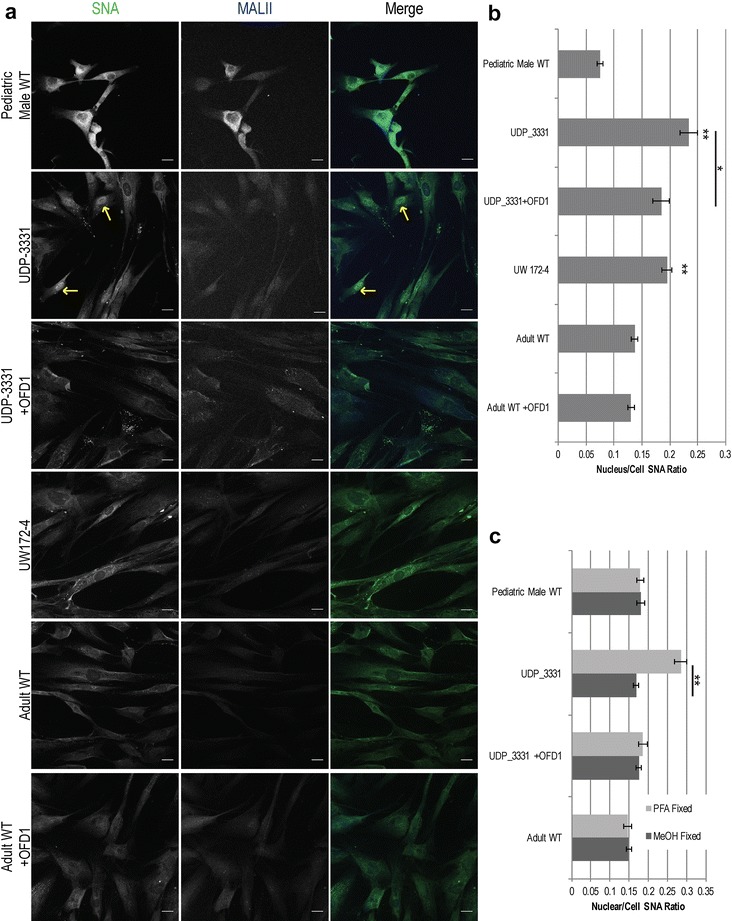
Glycome analyses of plasma and cultured dermal fibroblasts identified abnormal - and -linked glycosylation profiles. These findings replicated in two unrelated males with mutations. Cultured fibroblasts from affected individuals had a defect in ciliogenesis. The proband's fibroblasts also had an abnormally elevated nuclear sialylation signature and increased total cellular levels of CMP-sialic acid. Ciliogenesis and each glycosylation anomaly were rescued by expression of wild-type .
The rescue of ciliogenesis and glycosylation upon reintroduction of WT suggests that both contribute to the pathogenesis of JBTS10.
疾病发病机制的发现需要对可能失调的多个稳态过程进行系统的无偏筛选。我们在对一名患有10型乔布综合征(JBTS10)的5岁男孩的评估和诊断中阐述了这一原则。他携带OFD1突变p.Gln886Lysfs*2(NM_003611.2:c.2656del)并表现出乔布综合征的特征。
我们整合了外显子组测序、血浆和培养的皮肤成纤维细胞糖组的基质辅助激光解吸电离飞行时间质谱分析,以及对先证者的全面临床评估。通过免疫荧光进行纤毛形成和凝集素染色分析。使用高效阴离子交换色谱-脉冲安培检测法测量细胞核苷酸糖水平。统计分析采用学生氏检验和费舍尔精确检验。
血浆和培养的皮肤成纤维细胞的糖组分析确定了异常的O-连接和N-连接糖基化谱。这些发现在先证者的两个无血缘关系的男性突变体中得到了重复。来自受影响个体的培养成纤维细胞在纤毛发生方面存在缺陷。先证者的成纤维细胞还具有异常升高的核唾液酸化特征和CMP-唾液酸的总细胞水平增加。野生型OFD1的表达挽救了纤毛发生和每个糖基化异常。
重新引入野生型OFD1后纤毛发生和糖基化的挽救表明两者都有助于JBTS10的发病机制。