Zhang Chuanchao, Liu Juan, Shi Qianqian, Zeng Tao, Chen Luonan
State Key Laboratory of Software Engineering, School of Computer, Wuhan University, Wuhan, 430072, China.
Key Laboratory of Systems Biology, Innovation Center for Cell Signaling Network, Institute of Biochemistry and Cell Biology, Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences, Shanghai, 200031, China.
BMC Bioinformatics. 2017 Mar 14;18(Suppl 3):48. doi: 10.1186/s12859-017-1462-x.
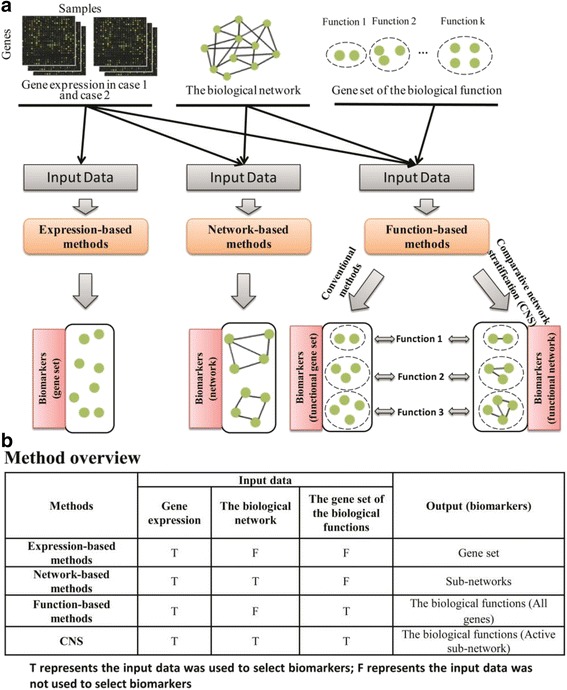
A major challenge of bioinformatics in the era of precision medicine is to identify the molecular biomarkers for complex diseases. It is a general expectation that these biomarkers or signatures have not only strong discrimination ability, but also readable interpretations in a biological sense. Generally, the conventional expression-based or network-based methods mainly capture differential genes or differential networks as biomarkers, however, such biomarkers only focus on phenotypic discrimination and usually have less biological or functional interpretation. Meanwhile, the conventional function-based methods could consider the biomarkers corresponding to certain biological functions or pathways, but ignore the differential information of genes, i.e., disregard the active degree of particular genes involved in particular functions, thereby resulting in less discriminative ability on phenotypes. Hence, it is strongly demanded to develop elaborate computational methods to directly identify functional network biomarkers with both discriminative power on disease states and readable interpretation on biological functions.
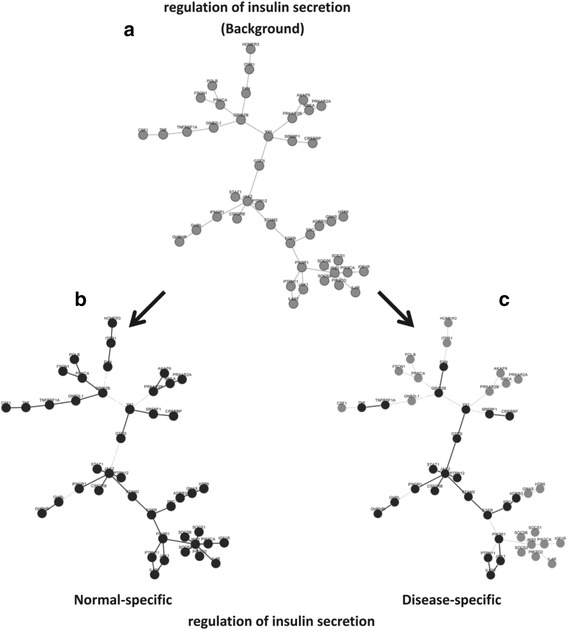
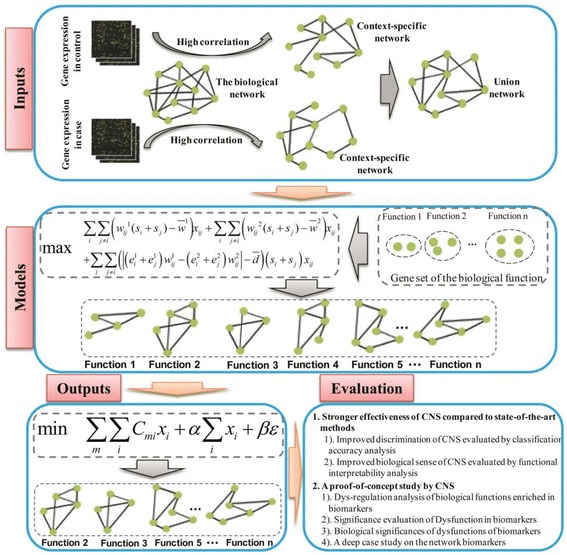
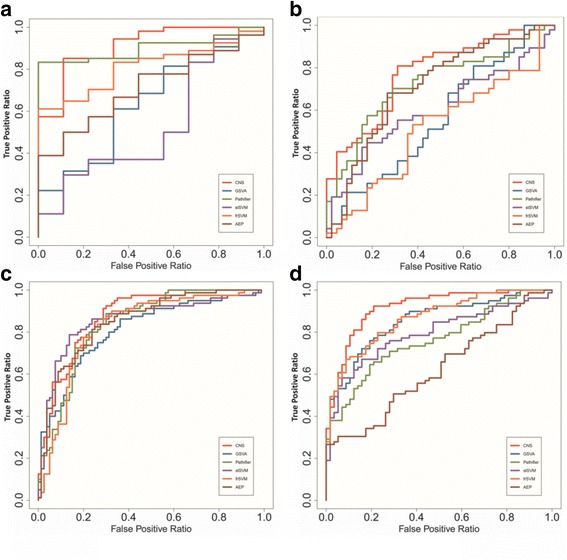
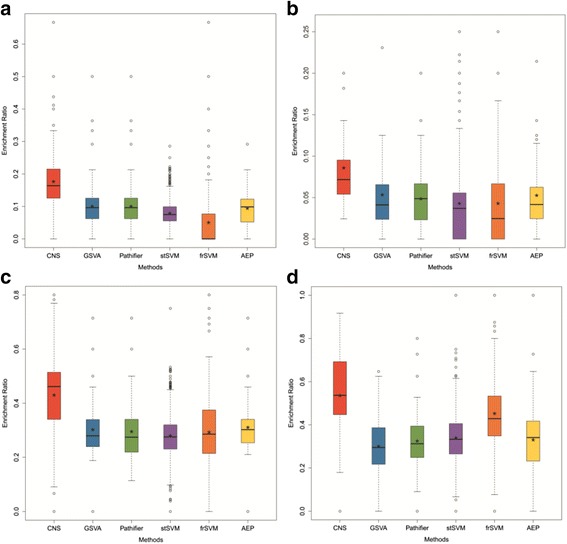
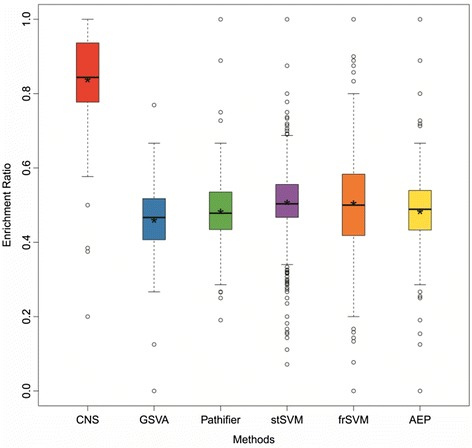
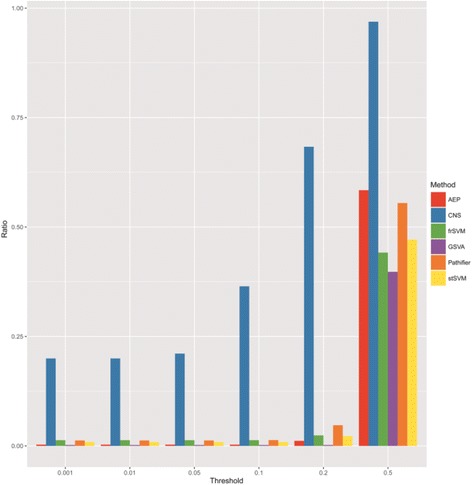
In this paper, we present a new computational framework based on an integer programming model, named as Comparative Network Stratification (CNS), to extract functional or interpretable network biomarkers, which are of strongly discriminative power on disease states and also readable interpretation on biological functions. In addition, CNS can not only recognize the pathogen biological functions disregarded by traditional Expression-based/Network-based methods, but also uncover the active network-structures underlying such dysregulated functions underestimated by traditional Function-based methods. To validate the effectiveness, we have compared CNS with five state-of-the-art methods, i.e. GSVA, Pathifier, stSVM, frSVM and AEP on four datasets of different complex diseases. The results show that CNS can enhance the discriminative power of network biomarkers, and further provide biologically interpretable information or disease pathogenic mechanism of these biomarkers. A case study on type 1 diabetes (T1D) demonstrates that CNS can identify many dysfunctional genes and networks previously disregarded by conventional approaches.
Therefore, CNS is actually a powerful bioinformatics tool, which can identify functional or interpretable network biomarkers with both discriminative power on disease states and readable interpretation on biological functions. CNS was implemented as a Matlab package, which is available at http://www.sysbio.ac.cn/cb/chenlab/images/CNSpackage_0.1.rar .
精准医学时代生物信息学的一个主要挑战是识别复杂疾病的分子生物标志物。人们普遍期望这些生物标志物或特征不仅具有强大的区分能力,而且在生物学意义上具有可读的解释。一般来说,传统的基于表达或基于网络的方法主要捕获差异基因或差异网络作为生物标志物,然而,这样的生物标志物只关注表型区分,通常生物学或功能解释较少。同时,传统的基于功能的方法可以考虑与某些生物学功能或途径相对应的生物标志物,但忽略了基因的差异信息,即忽视了参与特定功能的特定基因的活跃程度,从而导致对表型的区分能力较弱。因此,迫切需要开发精细的计算方法来直接识别对疾病状态具有区分能力且对生物学功能具有可读解释的功能网络生物标志物。
在本文中,我们提出了一种基于整数规划模型的新计算框架,名为比较网络分层(CNS),用于提取功能或可解释的网络生物标志物,这些生物标志物对疾病状态具有强大的区分能力,并且对生物学功能也具有可读的解释。此外,CNS不仅可以识别传统基于表达/基于网络的方法忽略的致病生物学功能,还可以揭示传统基于功能的方法低估的此类失调功能背后的活跃网络结构。为了验证其有效性,我们将CNS与五种最先进的方法,即GSVA、Pathifier、stSVM、frSVM和AEP,在四个不同复杂疾病的数据集上进行了比较。结果表明,CNS可以增强网络生物标志物的区分能力,并进一步提供这些生物标志物的生物学可解释信息或疾病致病机制。对1型糖尿病(T1D)的案例研究表明,CNS可以识别许多传统方法先前忽略的功能失调基因和网络。
因此,CNS实际上是一个强大的生物信息学工具,它可以识别对疾病状态具有区分能力且对生物学功能具有可读解释的功能或可解释网络生物标志物。CNS作为一个Matlab包实现,可在http://www.sysbio.ac.cn/cb/chenlab/images/CNSpackage_0.1.rar获取。