Fermo Elisa, Vercellati Cristina, Marcello Anna Paola, Zaninoni Anna, van Wijk Richard, Mirra Nadia, Curcio Cristina, Cortelezzi Agostino, Zanella Alberto, Barcellini Wilma, Bianchi Paola
U.O.C. Oncoematologia, U.O.S. Fisiopatologia delle Anemie, Fondazione IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milano, Italy.
Department of Clinical Chemistry and Haematology, University Medical Center Utrecht, Utrecht, Netherlands.
Case Rep Hematol. 2017;2017:2769570. doi: 10.1155/2017/2769570. Epub 2017 Mar 6.
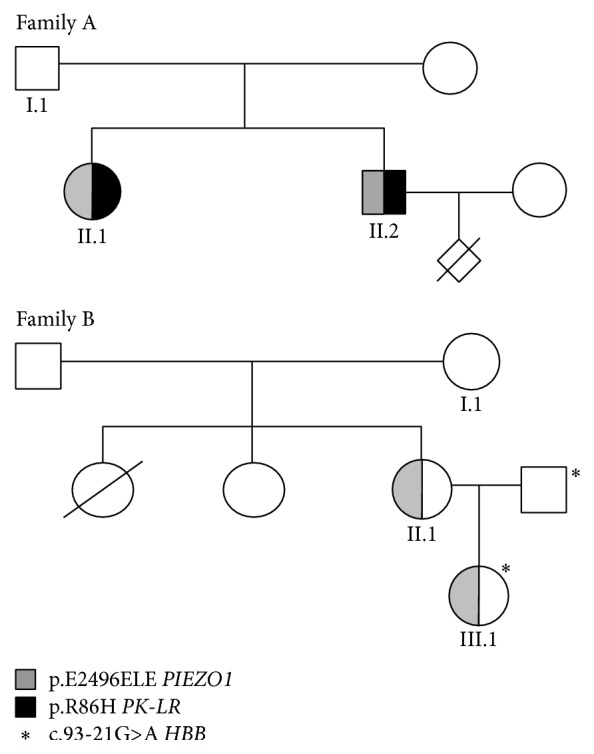
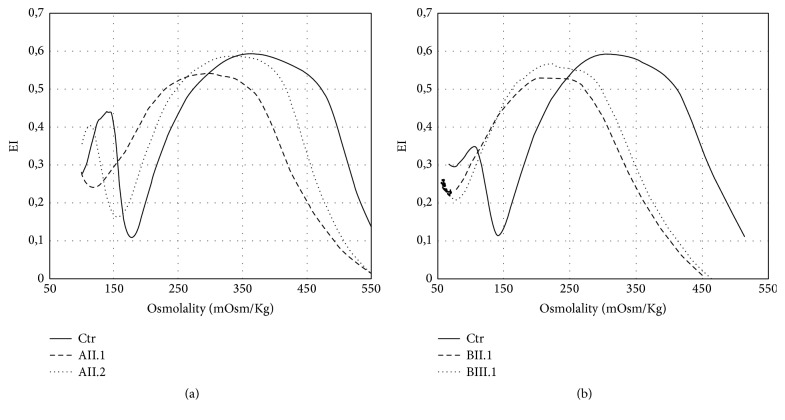
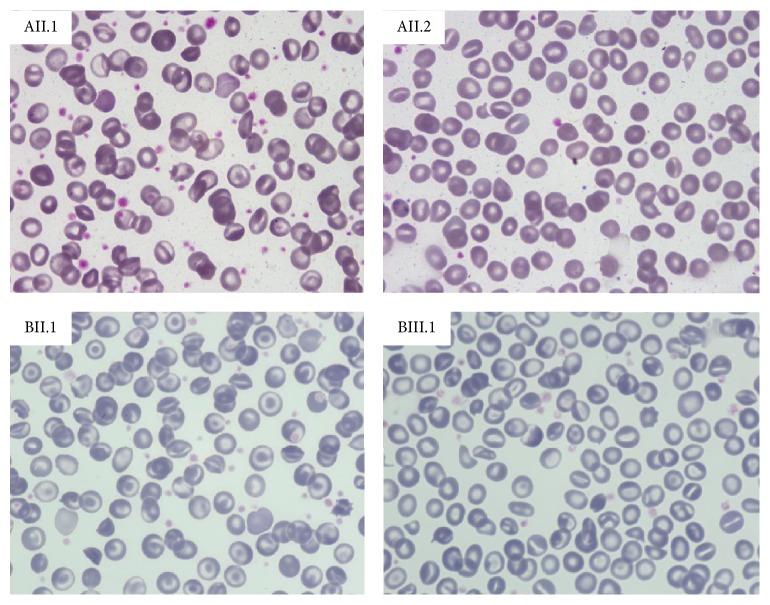
Hereditary xerocytosis (HX) is a rare disorder caused by defects of RBC permeability, associated with haemolytic anaemia of variable degree and iron overload. It is sometimes misdiagnosed as hereditary spherocytosis or other congenital haemolytic anaemia. Splenectomy is contraindicated due to increased risk of thromboembolic complications. We report the clinical, haematological, and molecular characteristics of four patients from two unrelated Italian families affected by HX, associated with beta-thalassemia trait and heterozygous pyruvate kinase deficiency, respectively. Two patients had been splenectomised and displayed thrombotic episodes. All patients had iron overload in the absence of transfusion, two of them requiring iron chelation. The diagnosis of HX was confirmed by LoRRca Osmoscan analysis showing a left-shifted curve. gene sequencing revealed the presence of mutation p.E2496ELE, showing that this is one of the most frequent mutations in this disease. The concomitant defects did not aggravate the clinical phenotype; however, in one patient, the initial diagnosis of pyruvate kinase deficiency delayed the correct diagnosis of HX for many years and resulted in splenectomy followed by thrombotic complications. The study underlines the importance of a precise diagnosis in HX, particularly in view of splenectomy, and the need of a molecular confirmation of suspected RBC enzymopathy.
遗传性口形红细胞增多症(HX)是一种由红细胞通透性缺陷引起的罕见疾病,与不同程度的溶血性贫血和铁过载相关。它有时会被误诊为遗传性球形红细胞增多症或其他先天性溶血性贫血。由于血栓栓塞并发症风险增加,脾切除术为禁忌。我们报告了来自两个不相关的意大利家庭的4例受HX影响患者的临床、血液学和分子特征,这4例患者分别合并β地中海贫血特征和杂合子丙酮酸激酶缺乏症。2例患者已接受脾切除术并出现血栓形成事件。所有患者在未输血情况下均有铁过载,其中2例需要进行铁螯合治疗。通过LoRRca Osmoscan分析显示曲线左移,确诊为HX。基因测序显示存在p.E2496ELE突变,表明这是该疾病最常见的突变之一。合并的缺陷并未加重临床表型;然而,在1例患者中,最初丙酮酸激酶缺乏症的诊断使HX的正确诊断延迟了多年,并导致脾切除术后出现血栓并发症。该研究强调了HX精确诊断的重要性,特别是考虑到脾切除术时,以及对疑似红细胞酶病进行分子确认的必要性。