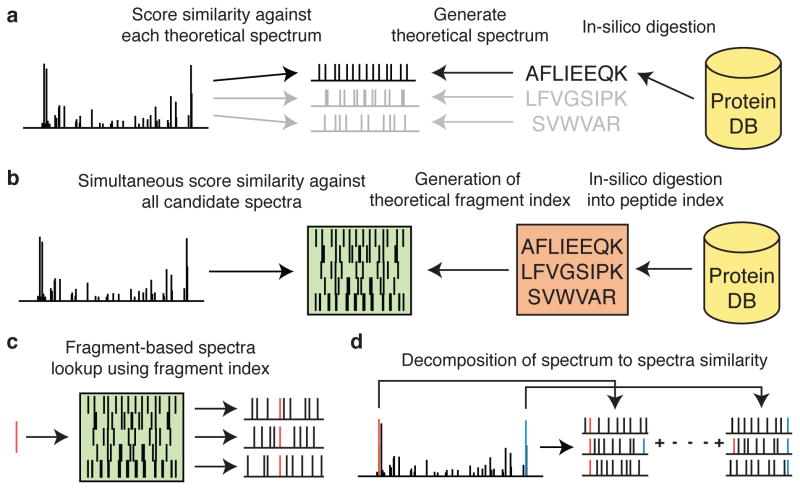
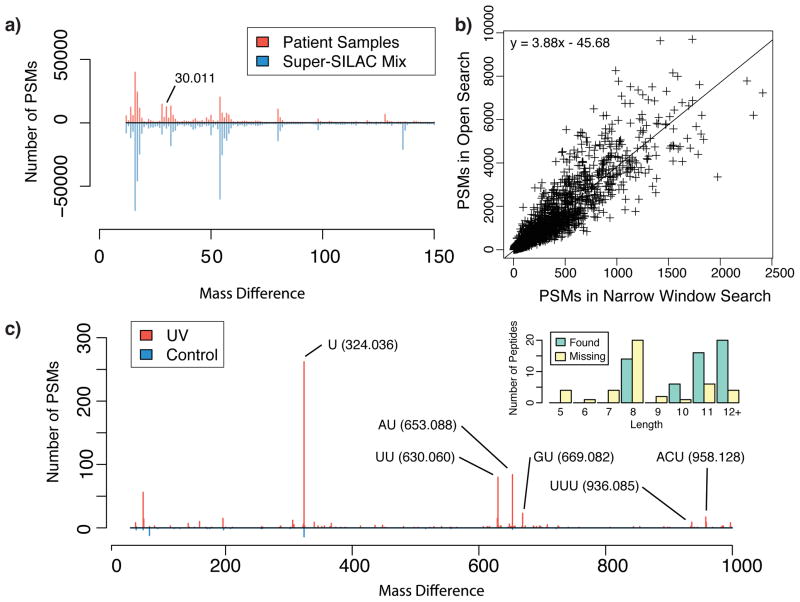
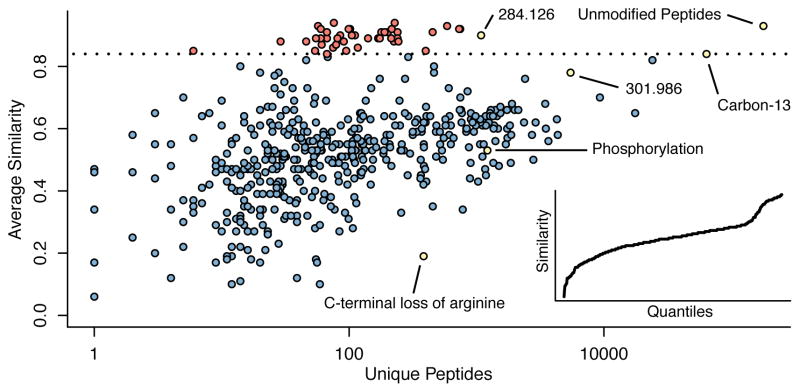
Kong Andy T, Leprevost Felipe V, Avtonomov Dmitry M, Mellacheruvu Dattatreya, Nesvizhskii Alexey I
Department of Computational Medicine and Bioinformatics, University of Michigan, Ann Arbor, Michigan, USA.
Department of Pathology, University of Michigan, Ann Arbor, Michigan, USA.
Nat Methods. 2017 May;14(5):513-520. doi: 10.1038/nmeth.4256. Epub 2017 Apr 10.
There is a need to better understand and handle the 'dark matter' of proteomics-the vast diversity of post-translational and chemical modifications that are unaccounted in a typical mass spectrometry-based analysis and thus remain unidentified. We present a fragment-ion indexing method, and its implementation in peptide identification tool MSFragger, that enables a more than 100-fold improvement in speed over most existing proteome database search tools. Using several large proteomic data sets, we demonstrate how MSFragger empowers the open database search concept for comprehensive identification of peptides and all their modified forms, uncovering dramatic differences in modification rates across experimental samples and conditions. We further illustrate its utility using protein-RNA cross-linked peptide data and using affinity purification experiments where we observe, on average, a 300% increase in the number of identified spectra for enriched proteins. We also discuss the benefits of open searching for improved false discovery rate estimation in proteomics.
有必要更好地理解和处理蛋白质组学中的“暗物质”——即翻译后修饰和化学修饰的巨大多样性,这些修饰在典型的基于质谱的分析中未被考虑到,因此仍然未被识别。我们提出了一种碎片离子索引方法及其在肽段鉴定工具MSFragger中的实现,该方法使速度比大多数现有的蛋白质组数据库搜索工具提高了100倍以上。使用几个大型蛋白质组数据集,我们展示了MSFragger如何支持开放数据库搜索概念,以全面鉴定肽段及其所有修饰形式,揭示不同实验样品和条件下修饰率的显著差异。我们进一步使用蛋白质-RNA交联肽数据和亲和纯化实验来说明其效用,在这些实验中,我们平均观察到富集蛋白质的鉴定谱图数量增加了300%。我们还讨论了开放搜索在蛋白质组学中改进错误发现率估计方面的好处。