Greenwald William W, Klitgord Niels, Seguritan Victor, Yooseph Shibu, Venter J Craig, Garner Chad, Nelson Karen E, Li Weizhong
Bioinformatics and Systems Biology, University of California San Diego, La Jolla, CA, USA.
Human Longevity Inc, San Diego, CA, USA.
BMC Genomics. 2017 Apr 13;18(1):296. doi: 10.1186/s12864-017-3679-5.
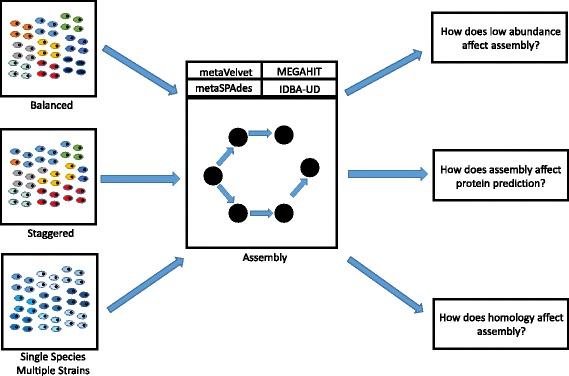
Metagenomics is the study of the microbial genomes isolated from communities found on our bodies or in our environment. By correctly determining the relation between human health and the human associated microbial communities, novel mechanisms of health and disease can be found, thus enabling the development of novel diagnostics and therapeutics. Due to the diversity of the microbial communities, strategies developed for aligning human genomes cannot be utilized, and genomes of the microbial species in the community must be assembled de novo. However, in order to obtain the best metagenomic assemblies, it is important to choose the proper assembler. Due to the rapidly evolving nature of metagenomics, new assemblers are constantly created, and the field has not yet agreed on a standardized process. Furthermore, the truth sets used to compare these methods are either too simple (computationally derived diverse communities) or complex (microbial communities of unknown composition), yielding results that are hard to interpret. In this analysis, we interrogate the strengths and weaknesses of five popular assemblers through the use of defined biological samples of known genomic composition and abundance. We assessed the performance of each assembler on their ability to reassemble genomes, call taxonomic abundances, and recreate open reading frames (ORFs).
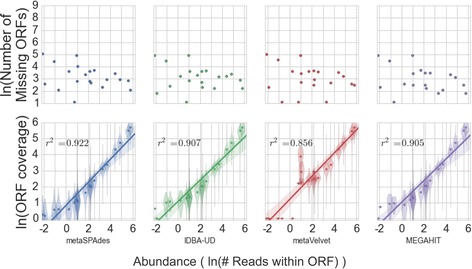
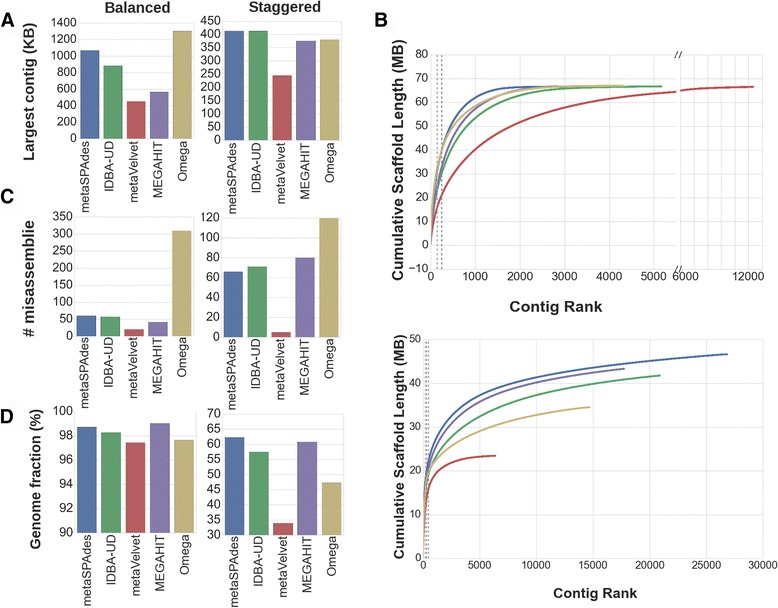
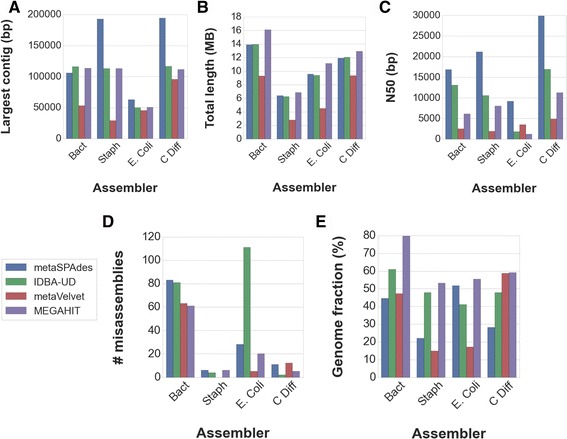
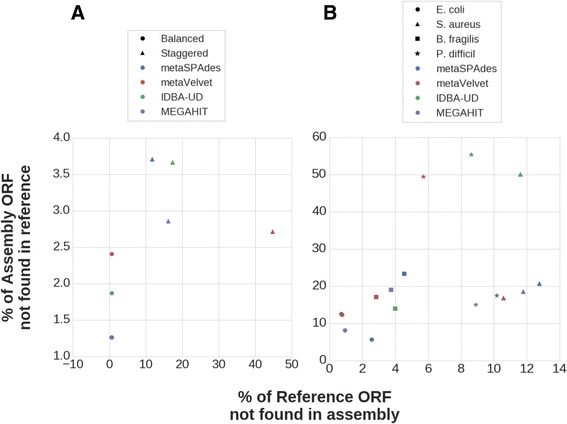
We tested five metagenomic assemblers: Omega, metaSPAdes, IDBA-UD, metaVelvet and MEGAHIT on known and synthetic metagenomic data sets. MetaSPAdes excelled in diverse sets, IDBA-UD performed well all around, metaVelvet had high accuracy in high abundance organisms, and MEGAHIT was able to accurately differentiate similar organisms within a community. At the ORF level, metaSPAdes and MEGAHIT had the least number of missing ORFs within diverse and similar communities respectively.
Depending on the metagenomics question asked, the correct assembler for the task at hand will differ. It is important to choose the appropriate assembler, and thus clearly define the biological problem of an experiment, as different assemblers will give different answers to the same question.
宏基因组学是对从我们身体上或环境中发现的群落中分离出的微生物基因组进行的研究。通过正确确定人类健康与人类相关微生物群落之间的关系,可以发现健康和疾病的新机制,从而推动新型诊断方法和治疗方法的开发。由于微生物群落的多样性,无法采用为比对人类基因组而开发的策略,必须从头组装群落中微生物物种的基因组。然而,为了获得最佳的宏基因组组装结果,选择合适的组装程序很重要。由于宏基因组学的快速发展,新的组装程序不断涌现,该领域尚未就标准化流程达成共识。此外,用于比较这些方法的真值集要么过于简单(通过计算得出的多样群落),要么过于复杂(组成未知的微生物群落),得出的结果难以解释。在本分析中,我们通过使用已知基因组组成和丰度的特定生物样本,探究了五种常用组装程序的优缺点。我们评估了每个组装程序在重新组装基因组、确定分类丰度以及重建开放阅读框(ORF)方面的能力。
我们在已知的和合成的宏基因组数据集上测试了五种宏基因组组装程序:Omega、metaSPAdes、IDBA-UD、metaVelvet和MEGAHIT。MetaSPAdes在多样数据集中表现出色,IDBA-UD整体表现良好,metaVelvet在高丰度生物体中具有较高的准确性,而MEGAHIT能够准确区分群落内的相似生物体。在ORF水平上,metaSPAdes和MEGAHIT分别在多样群落和相似群落中缺失的ORF数量最少。
根据所提出的宏基因组学问题,适用于手头任务的正确组装程序会有所不同。选择合适的组装程序很重要,因此要明确界定实验的生物学问题,因为不同的组装程序对同一问题会给出不同的答案。