van der Walt Andries Johannes, van Goethem Marc Warwick, Ramond Jean-Baptiste, Makhalanyane Thulani Peter, Reva Oleg, Cowan Don Arthur
Centre for Microbial Ecology and Genomics (CMEG), Department of Genetics, University of Pretoria, Natural Sciences Building 2, Lynnwood Road, Pretoria, 0028, South Africa.
Centre for Bioinformatics and Computational Biology, Department of Biochemistry, University of Pretoria, Pretoria, South Africa.
BMC Genomics. 2017 Jul 10;18(1):521. doi: 10.1186/s12864-017-3918-9.
Metagenomics allows unprecedented access to uncultured environmental microorganisms. The analysis of metagenomic sequences facilitates gene prediction and annotation, and enables the assembly of draft genomes, including uncultured members of a community. However, while several platforms have been developed for this critical step, there is currently no clear framework for the assembly of metagenomic sequence data.
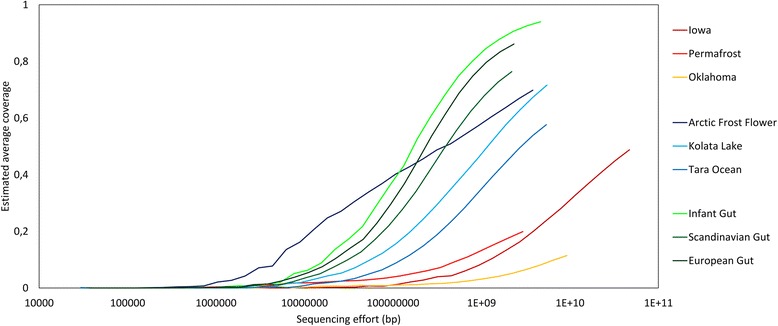
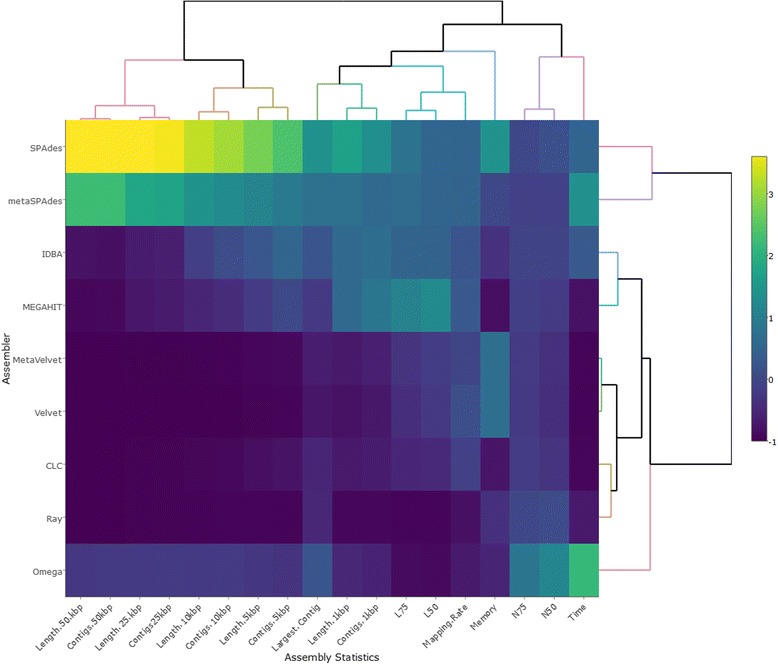
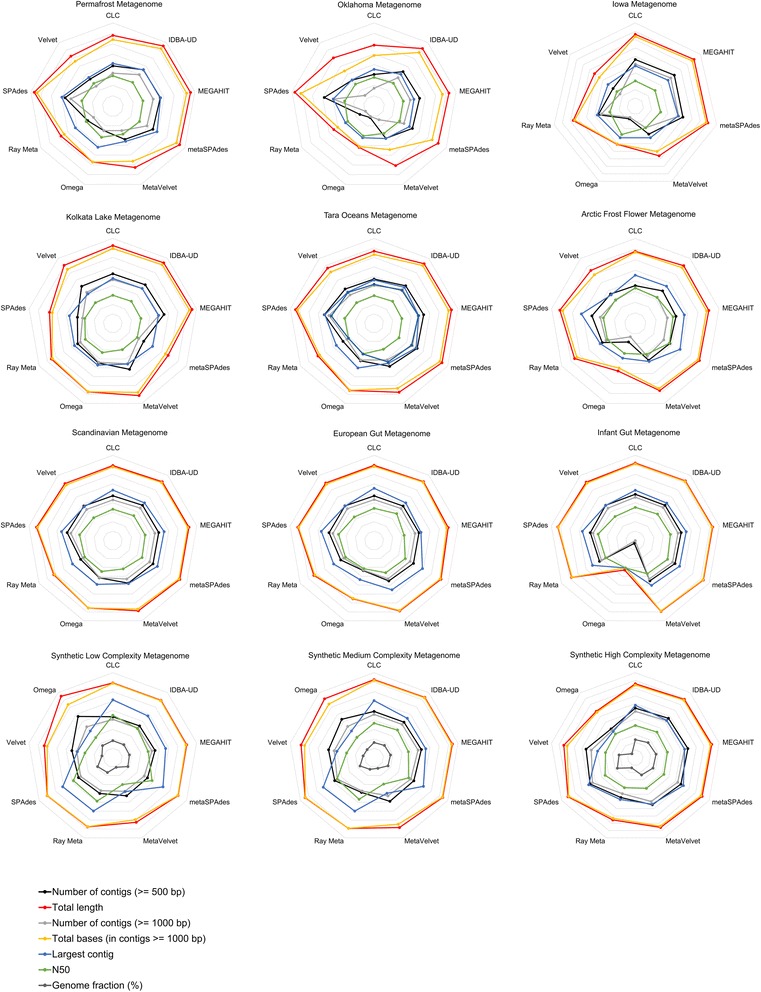
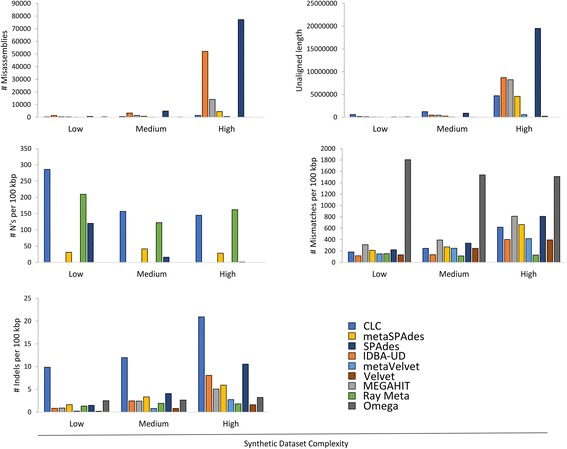
To assist with selection of an appropriate metagenome assembler we evaluated the capabilities of nine prominent assembly tools on nine publicly-available environmental metagenomes, as well as three simulated datasets. Overall, we found that SPAdes provided the largest contigs and highest N50 values across 6 of the 9 environmental datasets, followed by MEGAHIT and metaSPAdes. MEGAHIT emerged as a computationally inexpensive alternative to SPAdes, assembling the most complex dataset using less than 500 GB of RAM and within 10 hours.
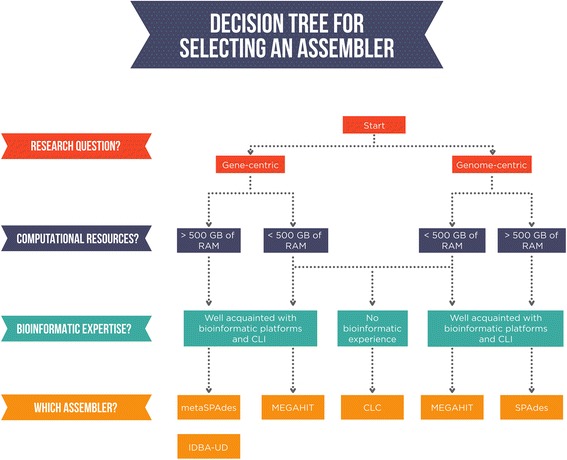
We found that assembler choice ultimately depends on the scientific question, the available resources and the bioinformatic competence of the researcher. We provide a concise workflow for the selection of the best assembly tool.
宏基因组学使人们能够以前所未有的方式研究未培养的环境微生物。宏基因组序列分析有助于基因预测和注释,并能够组装基因组草图,包括群落中未培养的成员。然而,尽管已经开发了多个平台用于这一关键步骤,但目前尚无用于宏基因组序列数据组装的明确框架。
为了帮助选择合适的宏基因组组装工具,我们在9个公开可用的环境宏基因组以及3个模拟数据集上评估了9种著名组装工具的性能。总体而言,我们发现SPAdes在9个环境数据集中的6个上产生了最长的重叠群和最高的N50值,其次是MEGAHIT和metaSPAdes。MEGAHIT是一种计算成本较低的替代SPAdes的工具,它使用不到500GB的随机存取存储器在10小时内组装了最复杂的数据集。
我们发现组装工具的选择最终取决于科学问题、可用资源以及研究人员的生物信息学能力。我们提供了一个选择最佳组装工具的简明工作流程。