Zhou Qinghong, Zhou Can, Zheng Wei, Mason Annaliese S, Fan Shuying, Wu Caijun, Fu Donghui, Huang Yingjin
Key Laboratory of Crop Physiology, Ecology and Genetic Breeding, Ministry of Education, Agronomy College, Jiangxi Agricultural UniversityNanchang, China.
Jiangxi Institute of Red SoilJinxian, China.
Front Plant Sci. 2017 Apr 28;8:648. doi: 10.3389/fpls.2017.00648. eCollection 2017.
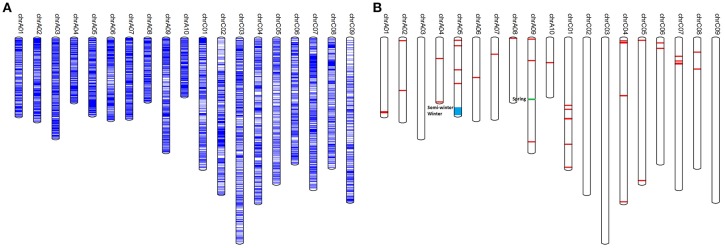
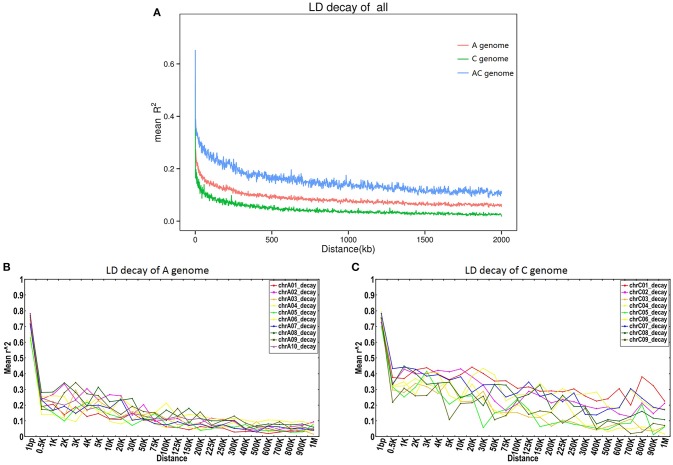
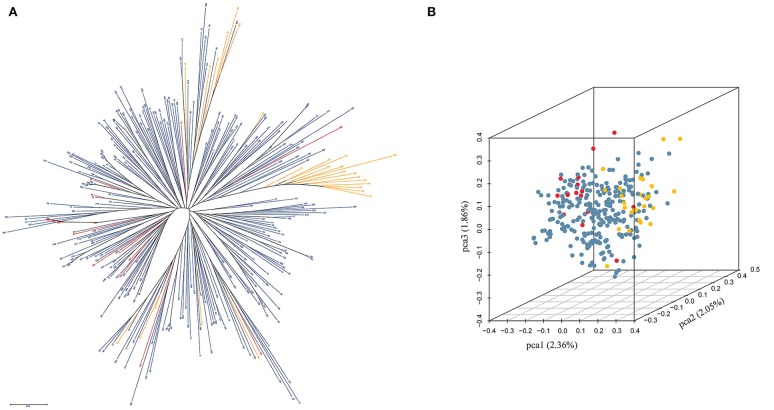
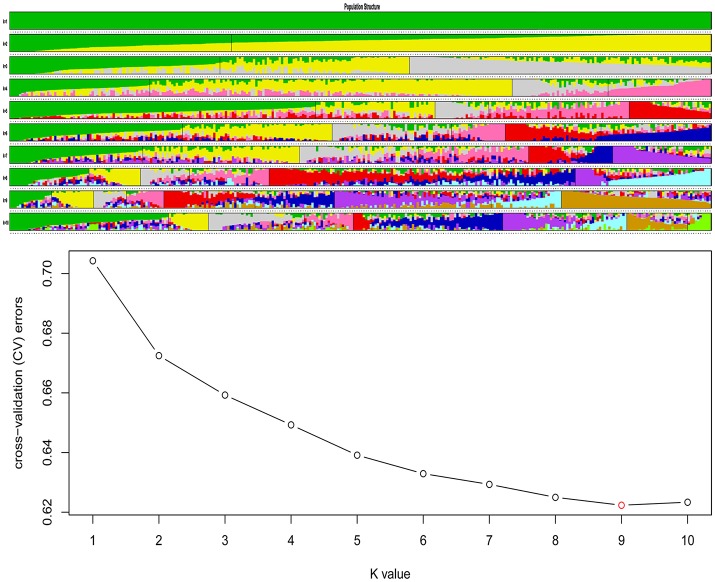
Single Nucleotide Polymorphisms (SNPs) are the most abundant and richest form of genomic polymorphism, and hence make highly favorable markers for genetic map construction and genome-wide association studies. In this study, a total of 300 rapeseed accessions (278 representative of Chinese germplasm, plus 22 outgroup accessions of different origins and ecotypes) were collected and sequenced using Specific-Locus Amplified Fragment Sequencing (SLAF-seq) technology, obtaining 660.25M reads with an average sequencing depth of 6.27 × and a mean Q30 of 85.96%. Based on the 238,711 polymorphic SLAF tags a total of 1,197,282 SNPs were discovered, and a subset of 201,817 SNPs with minor allele frequency >0.05 and integrity >0.8 were selected. Of these, 30,877 were designated SNP "hotspots," and 41 SNP-rich genomic regions could be delineated, with 100 genes associated with plant resistance, vernalization response, and signal transduction detected in these regions. Subsequent analysis of genetic diversity, linkage disequilibrium (LD), and population structure in the 300 accessions was carried out based on the 201,817 SNPs. Nine subpopulations were observed based on the population structure analysis. Hierarchical clustering and principal component analysis divided the 300 varieties roughly in accordance with their ecotype origins. However, spring-type varieties were intermingled with semi-winter type varieties, indicating frequent hybridization between spring and semi-winter ecotypes in China. In addition, LD decay across the whole genome averaged 299 kb when = 0.1, but the LD decay in the A genome (43 kb) was much shorter than in the C genome (1,455 kb), supporting the targeted introgression of the A genome from progenitor species into Chinese rapeseed. This study also lays the foundation for genetic analysis of important agronomic traits using this rapeseed population.
单核苷酸多态性(SNPs)是基因组多态性中最丰富、最常见的形式,因此是遗传图谱构建和全基因组关联研究的理想标记。本研究共收集了300份油菜种质(278份代表中国种质,外加22份不同来源和生态型的外类群种质),并采用特异位点扩增片段测序(SLAF-seq)技术进行测序,获得660.25M条reads,平均测序深度为6.27×,平均Q30为85.96%。基于238,711个多态性SLAF标签,共发现1,197,282个SNPs,并选择了201,817个次要等位基因频率>0.05且完整性>0.8的SNPs子集。其中,30,877个被指定为SNP“热点”,并划定了41个富含SNP的基因组区域,在这些区域中检测到100个与植物抗性、春化反应和信号转导相关的基因。随后基于这201,817个SNPs对300份种质进行了遗传多样性、连锁不平衡(LD)和群体结构分析。基于群体结构分析观察到9个亚群。层次聚类和主成分分析将300个品种大致按照其生态型来源进行划分。然而,春性品种与半冬性品种相互混杂,表明中国春性和半冬性生态型之间频繁杂交。此外,当r = 0.1时,全基因组的LD衰减平均为299 kb,但A基因组中的LD衰减(43 kb)比C基因组中的LD衰减(1,455 kb)短得多,支持从祖先物种向中国油菜定向导入A基因组。本研究也为利用该油菜群体对重要农艺性状进行遗传分析奠定了基础。