Banerjee Sudip, Melnyk Stepan B, Krager Kimberly J, Aykin-Burns Nukhet, McCullough Sandra S, James Laura P, Hinson Jack A
Departments of Pharmacology and Toxicology (SB, LPJ, JAH), Pediatrics (SBM, SSM, LPJ), and Pharmaceutical Sciences (KJK, NA-B), University of Arkansas for Medical Sciences, and Arkansas Children's Hospital Research Institute, Little Rock, AR, 72205.
Toxicol Rep. 2017;4:134-142. doi: 10.1016/j.toxrep.2017.02.005.
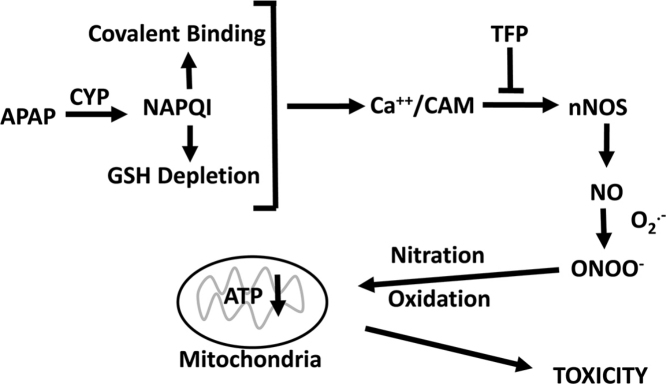
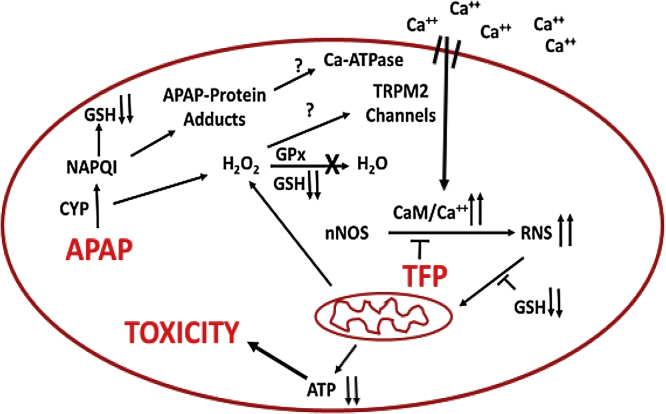
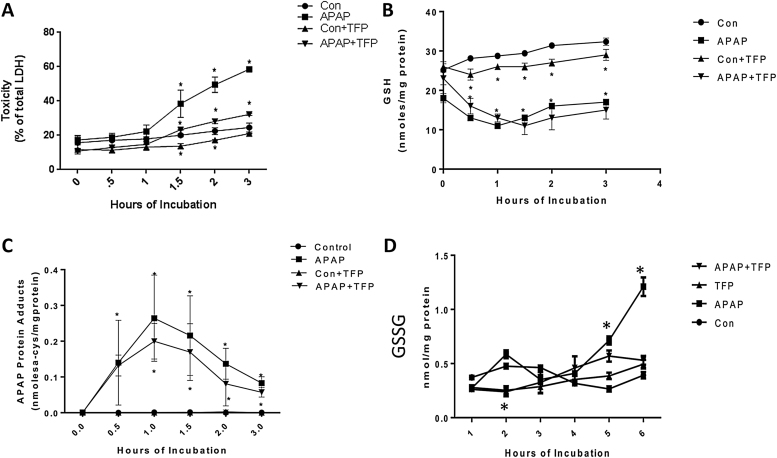
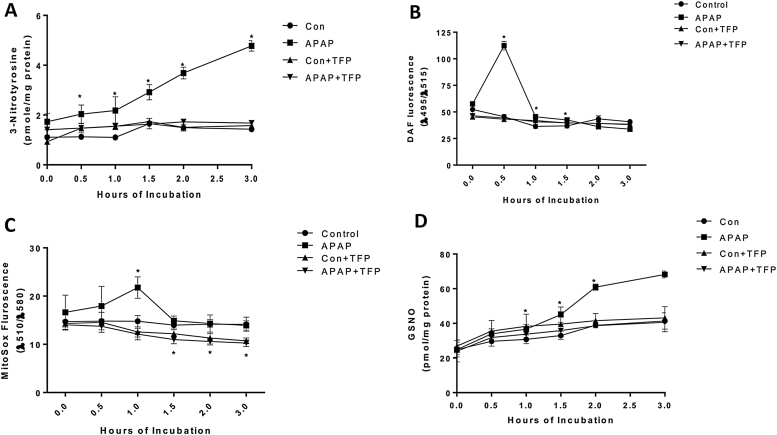
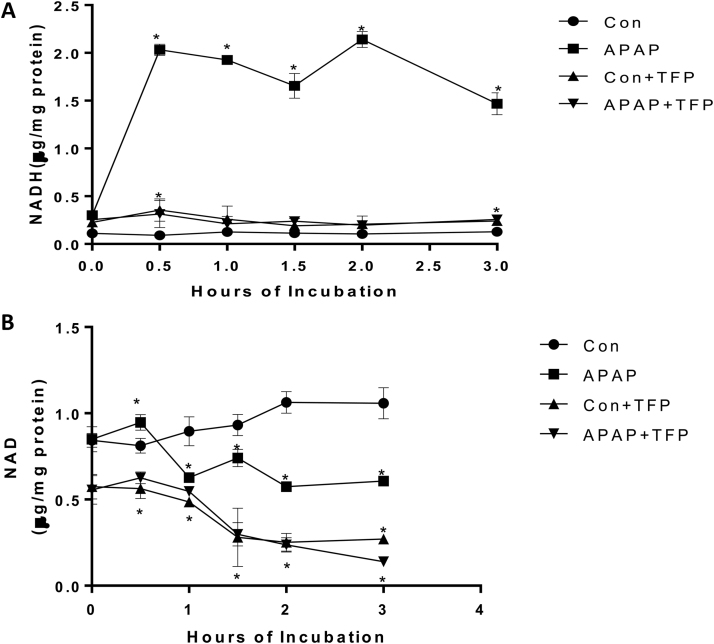
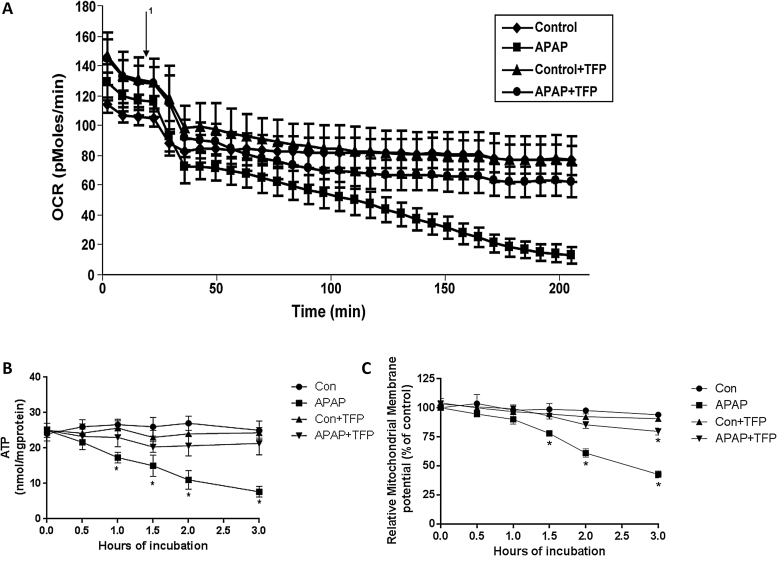
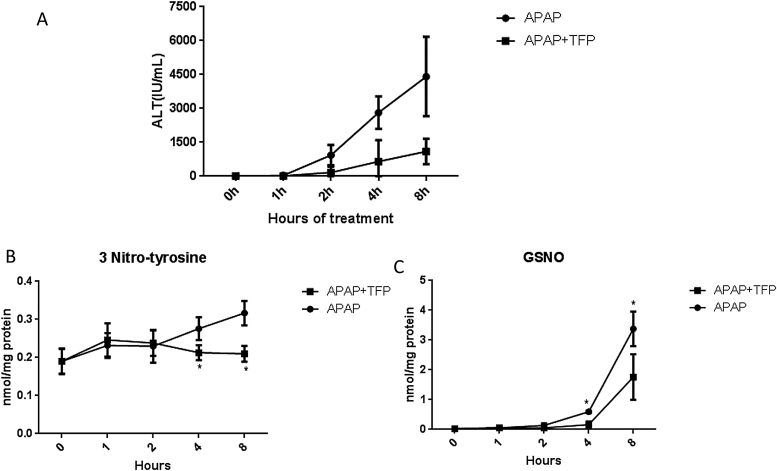
The hepatotoxicity of acetaminophen (APAP) occurs by initial metabolism to N-acetyl-p-benzoquinone imine which depletes GSH and forms APAP-protein adducts. Subsequently, the reactive nitrogen species peroxynitrite is formed from nitric oxide (NO) and superoxide leading to 3-nitrotyrosine in proteins. Toxicity occurs with inhibited mitochondrial function. We previously reported that in hepatocytes the nNOS (NOS1) inhibitor NANT inhibited APAP toxicity, reactive nitrogen and oxygen species formation, and mitochondrial dysfunction. In this work we examined the effect of trifluoperazine (TFP), a calmodulin antagonist that inhibits calcium induced nNOS activation, on APAP hepatotoxicity and reactive nitrogen formation in murine hepatocytes and . In freshly isolated hepatocytes TFP inhibited APAP induced toxicity, reactive nitrogen formation (NO, GSNO, and 3-nitrotyrosine in protein), reactive oxygen formation (superoxide), loss of mitochondrial membrane potential, decreased ATP production, decreased oxygen consumption rate, and increased NADH accumulation. TFP did not alter APAP induced GSH depletion in the hepatocytes or the formation of APAP protein adducts which indicated that reactive metabolite formation was not inhibited. Since we previously reported that TFP inhibits the hepatotoxicity of APAP in mice without altering hepatic APAP-protein adduct formation, we examined the APAP treated mouse livers for evidence of reactive nitrogen formation. 3-Nitrotyrosine in hepatic proteins and GSNO were significantly increased in APAP treated mouse livers and decreased in the livers of mice treated with APAP plus TFP. These data are consistent with a hypothesis that APAP hepatotoxicity occurs with altered calcium metabolism, activation of nNOS leading to increased reactive nitrogen formation, and mitochondrial dysfunction.
对乙酰氨基酚(APAP)的肝毒性最初是通过代谢生成N - 乙酰 - 对苯醌亚胺,该物质会消耗谷胱甘肽(GSH)并形成APAP - 蛋白质加合物。随后,活性氮物质过氧亚硝酸盐由一氧化氮(NO)和超氧化物形成,导致蛋白质中出现3 - 硝基酪氨酸。毒性伴随着线粒体功能的抑制而发生。我们之前报道过,在肝细胞中,nNOS(一氧化氮合酶1)抑制剂NANT可抑制APAP毒性、活性氮和氧物质的形成以及线粒体功能障碍。在这项研究中,我们检测了三氟拉嗪(TFP),一种抑制钙诱导的nNOS激活的钙调蛋白拮抗剂,对小鼠肝细胞中APAP肝毒性和活性氮形成的影响。在新鲜分离的肝细胞中,TFP抑制了APAP诱导的毒性、活性氮形成(蛋白质中的NO、GSNO和3 - 硝基酪氨酸)、活性氧形成(超氧化物)、线粒体膜电位丧失、ATP生成减少、氧消耗率降低以及NADH积累增加。TFP并未改变APAP诱导的肝细胞中GSH消耗或APAP蛋白质加合物的形成,这表明活性代谢物的形成未受抑制。由于我们之前报道过TFP可抑制小鼠中APAP的肝毒性而不改变肝脏中APAP - 蛋白质加合物的形成,我们检测了经APAP处理的小鼠肝脏中活性氮形成的证据。在经APAP处理的小鼠肝脏中,肝蛋白质中的3 - 硝基酪氨酸和GSNO显著增加,而在经APAP加TFP处理的小鼠肝脏中则减少。这些数据与一个假设一致,即APAP肝毒性的发生与钙代谢改变、nNOS激活导致活性氮形成增加以及线粒体功能障碍有关。