Department of Computer Sciences.
Department of Biostatistics and Medical Informatics, University of Wisconsin, Madison, WI 53706, USA.
Bioinformatics. 2017 Sep 15;33(18):2914-2923. doi: 10.1093/bioinformatics/btx334.
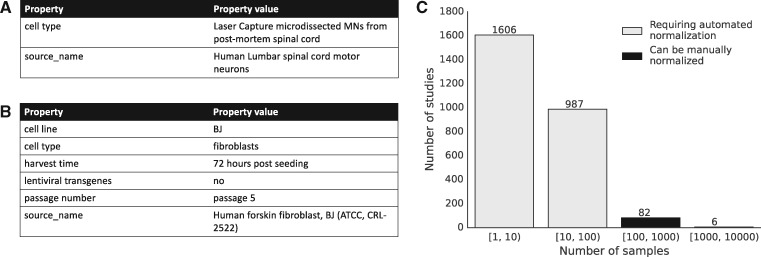
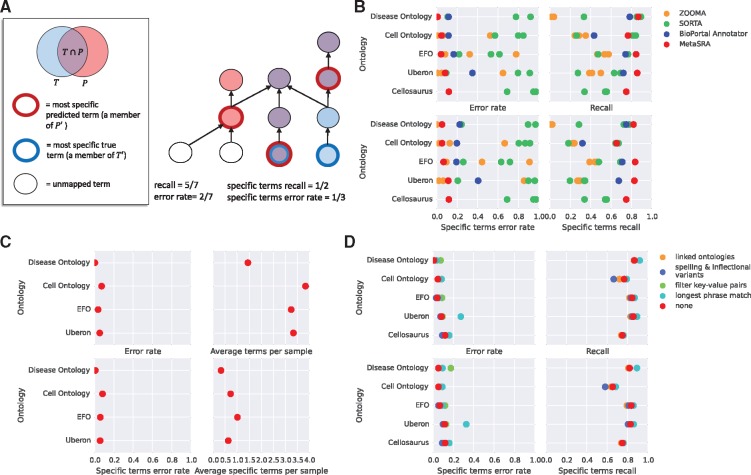
The NCBI's Sequence Read Archive (SRA) promises great biological insight if one could analyze the data in the aggregate; however, the data remain largely underutilized, in part, due to the poor structure of the metadata associated with each sample. The rules governing submissions to the SRA do not dictate a standardized set of terms that should be used to describe the biological samples from which the sequencing data are derived. As a result, the metadata include many synonyms, spelling variants and references to outside sources of information. Furthermore, manual annotation of the data remains intractable due to the large number of samples in the archive. For these reasons, it has been difficult to perform large-scale analyses that study the relationships between biomolecular processes and phenotype across diverse diseases, tissues and cell types present in the SRA.
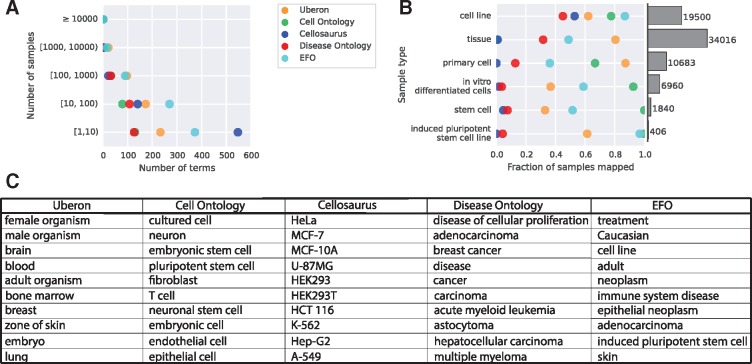
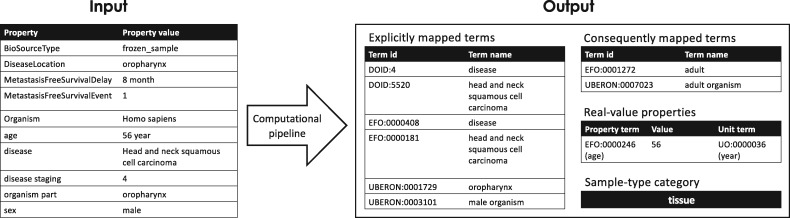
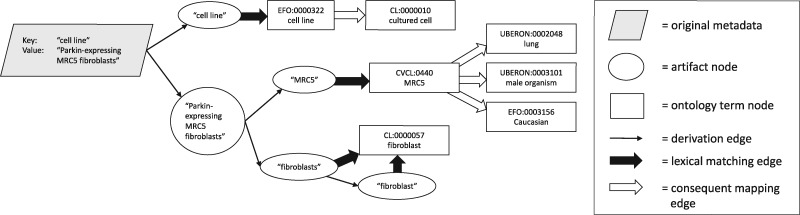
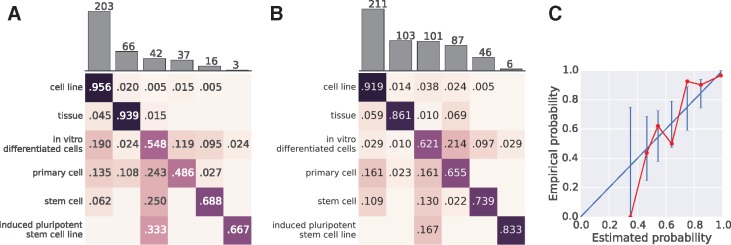
We present MetaSRA, a database of normalized SRA human sample-specific metadata following a schema inspired by the metadata organization of the ENCODE project. This schema involves mapping samples to terms in biomedical ontologies, labeling each sample with a sample-type category, and extracting real-valued properties. We automated these tasks via a novel computational pipeline.
The MetaSRA is available at metasra.biostat.wisc.edu via both a searchable web interface and bulk downloads. Software implementing our computational pipeline is available at http://github.com/deweylab/metasra-pipeline.
Supplementary data are available at Bioinformatics online.
NCBI 的序列读取档案 (SRA) 承诺,如果能够对数据进行汇总分析,将提供巨大的生物学见解;然而,由于与每个样本相关的元数据结构较差,这些数据在很大程度上仍未得到充分利用。提交给 SRA 的规则并没有规定应该使用一组标准化的术语来描述测序数据所源自的生物样本。因此,元数据包括许多同义词、拼写变体和对外部信息源的引用。此外,由于档案中样本数量众多,数据的手动注释仍然难以处理。由于这些原因,很难进行大规模的分析,这些分析研究了 SRA 中存在的不同疾病、组织和细胞类型之间生物分子过程和表型之间的关系。
我们提出了 MetaSRA,这是一个经过规范化的 SRA 人类样本特定元数据数据库,其模式受到 ENCODE 项目元数据组织的启发。该模式涉及将样本映射到生物医学本体论中的术语,用样本类型类别标记每个样本,并提取实值属性。我们通过一个新颖的计算管道自动执行这些任务。
MetaSRA 可通过可搜索的网络界面和批量下载在 metasra.biostat.wisc.edu 上获得。实现我们的计算管道的软件可在 http://github.com/deweylab/metasra-pipeline 上获得。
补充数据可在 Bioinformatics 在线获得。