Ohta Tazro, Nakazato Takeru, Bono Hidemasa
Gigascience. 2017 Jun 1;6(6):1-8. doi: 10.1093/gigascience/gix029.
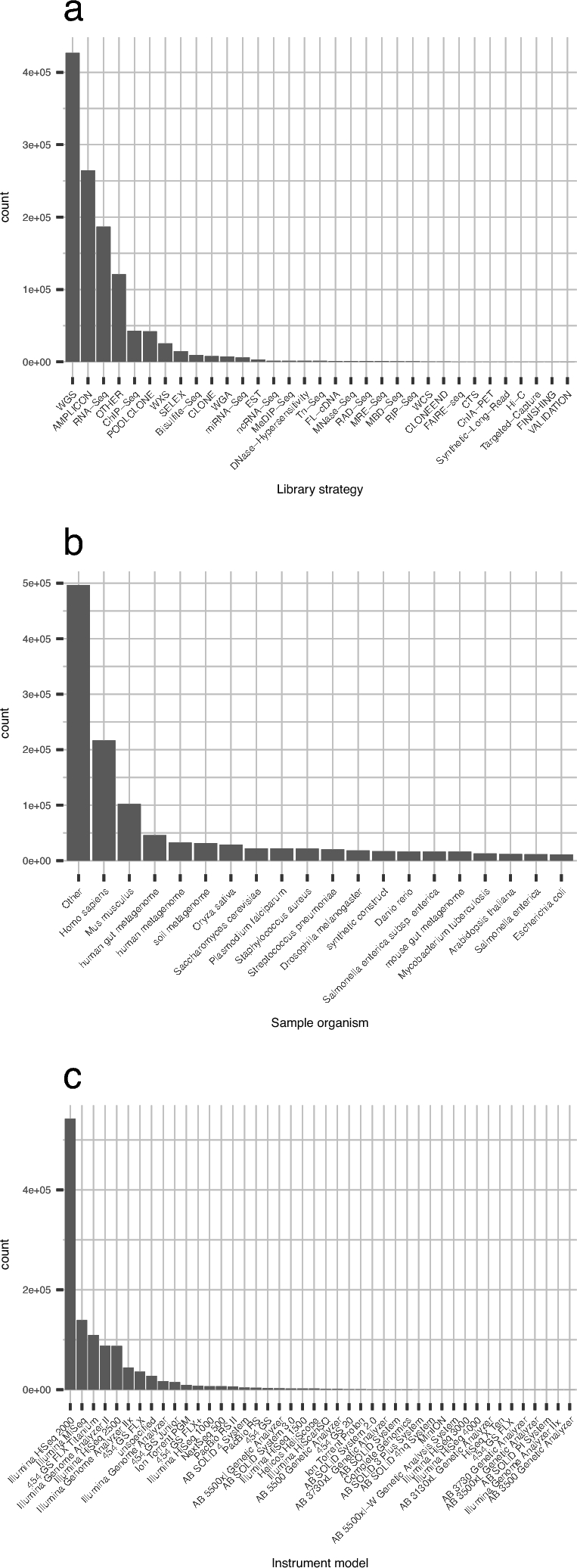
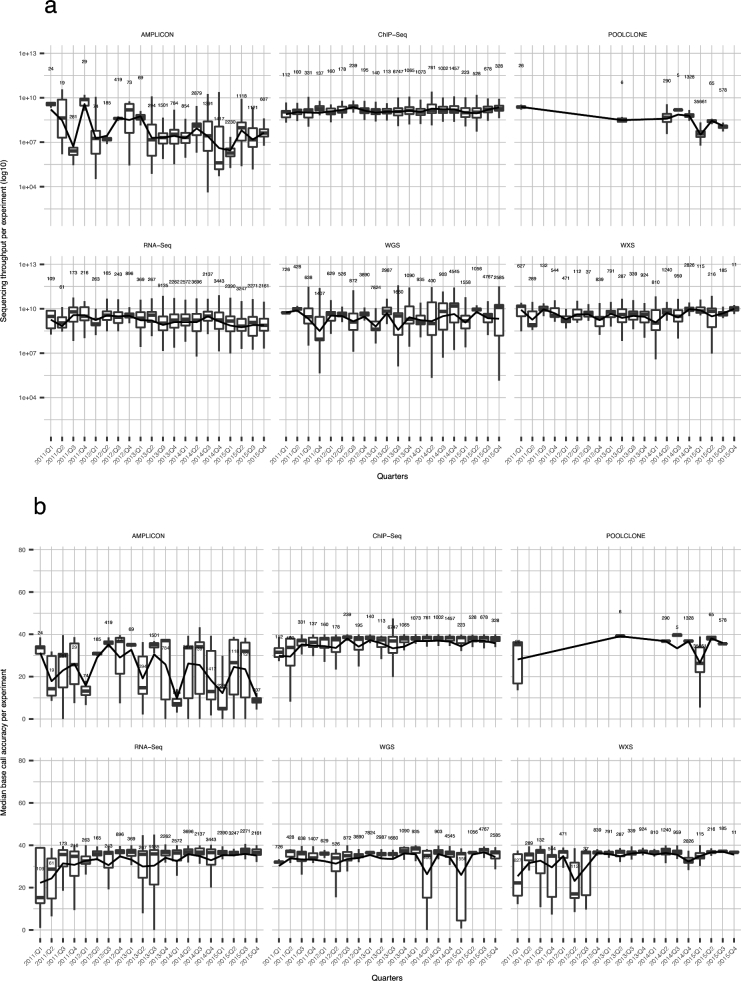
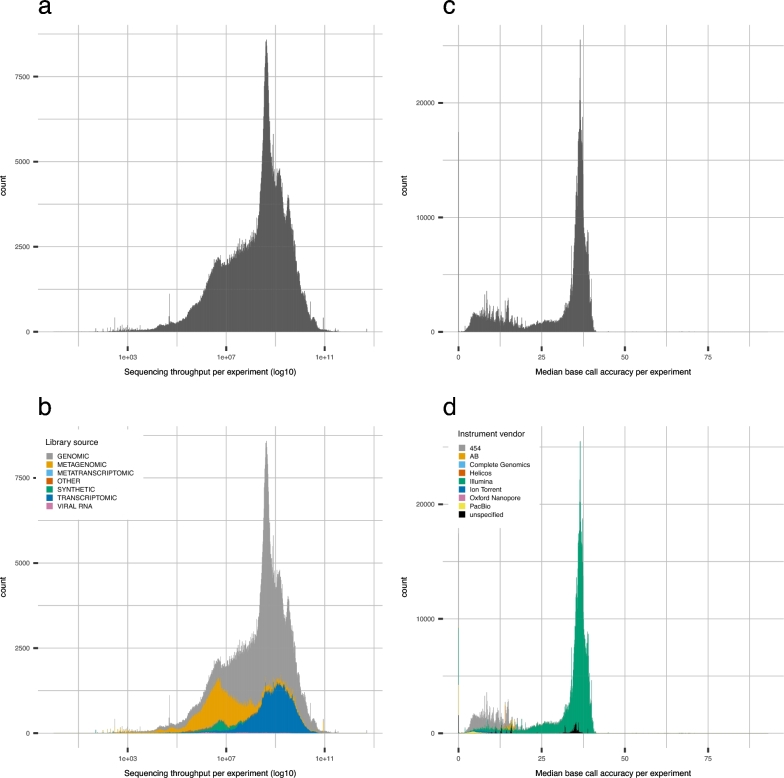
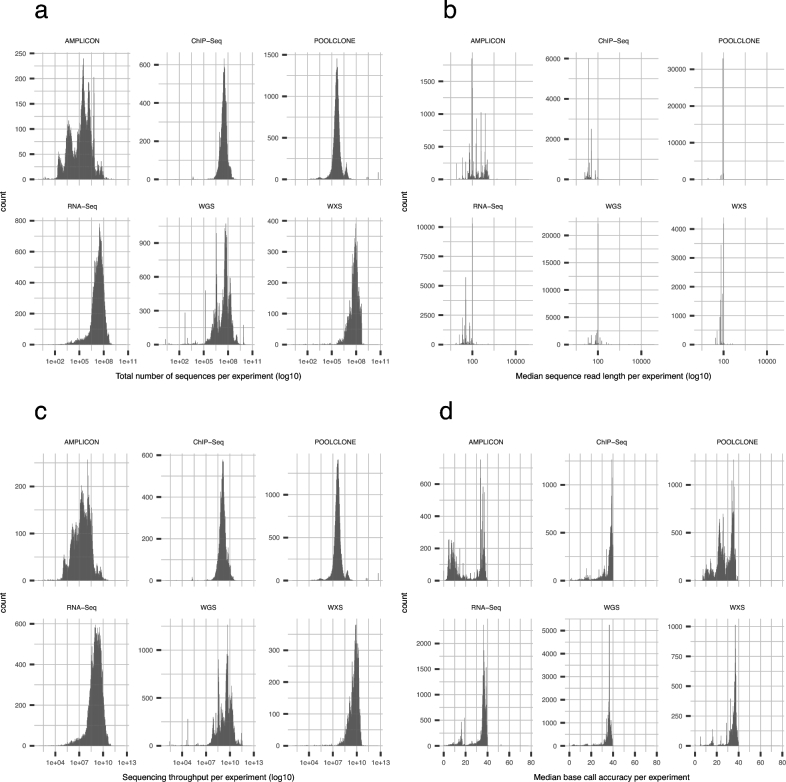
It is important for public data repositories to promote the reuse of archived data. In the growing field of omics science, however, the increasing number of submissions of high-throughput sequencing (HTSeq) data to public repositories prevents users from choosing a suitable data set from among the large number of search results. Repository users need to be able to set a threshold to reduce the number of results to obtain a suitable subset of high-quality data for reanalysis. We calculated the quality of sequencing data archived in a public data repository, the Sequence Read Archive (SRA), by using the quality control software FastQC. We obtained quality values for 1 171 313 experiments, which can be used to evaluate the suitability of data for reuse. We also visualized the data distribution in SRA by integrating the quality information and metadata of experiments and samples. We provide quality information of all of the archived sequencing data, which enable users to obtain sufficient quality sequencing data for reanalyses. The calculated quality data are available to the public in various formats. Our data also provide an example of enhancing the reuse of public data by adding metadata to published research data by a third party.
对于公共数据存储库而言,促进存档数据的再利用非常重要。然而,在不断发展的组学科学领域,向公共存储库提交的高通量测序(HTSeq)数据数量日益增加,这使得用户难以从大量搜索结果中选择合适的数据集。存储库用户需要能够设置一个阈值,以减少结果数量,从而获得用于重新分析的高质量数据的合适子集。我们使用质量控制软件FastQC计算了公共数据存储库序列读取存档(SRA)中存档的测序数据质量。我们获得了1171313个实验的质量值,这些值可用于评估数据再利用的适用性。我们还通过整合实验和样本的质量信息与元数据,直观展示了SRA中的数据分布。我们提供了所有存档测序数据的质量信息,这使用户能够获取足够高质量的测序数据用于重新分析。计算出的质量数据以各种格式向公众提供。我们的数据还提供了一个通过第三方为已发表的研究数据添加元数据来提高公共数据再利用的示例。