Wang Zhuo, Jin Shuilin, Liu Guiyou, Zhang Xiurui, Wang Nan, Wu Deliang, Hu Yang, Zhang Chiping, Jiang Qinghua, Xu Li, Wang Yadong
Department of Mathematics, Harbin Institute of Technology, Harbin, Heilongjiang, 150001, West Dazhi Street, China.
School of Computer Science and Technology, Harbin Institute of Technology, Harbin, Heilongjiang, 150001, West Dazhi Street, China.
BMC Bioinformatics. 2017 May 23;18(1):270. doi: 10.1186/s12859-017-1647-3.
The development of single-cell RNA sequencing has enabled profound discoveries in biology, ranging from the dissection of the composition of complex tissues to the identification of novel cell types and dynamics in some specialized cellular environments. However, the large-scale generation of single-cell RNA-seq (scRNA-seq) data collected at multiple time points remains a challenge to effective measurement gene expression patterns in transcriptome analysis.
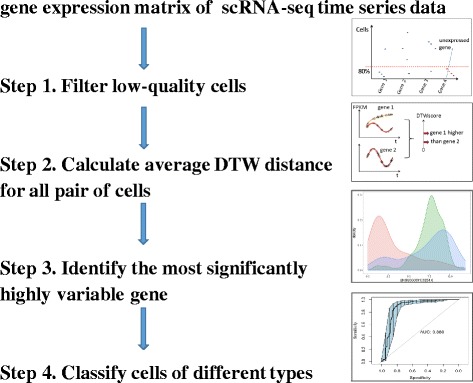
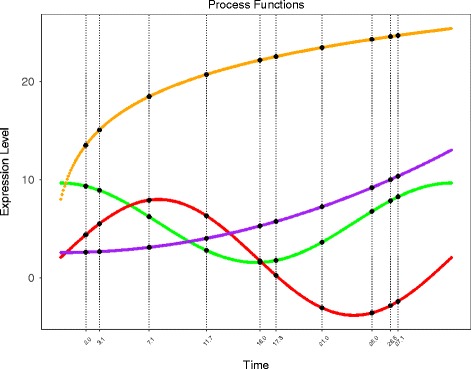
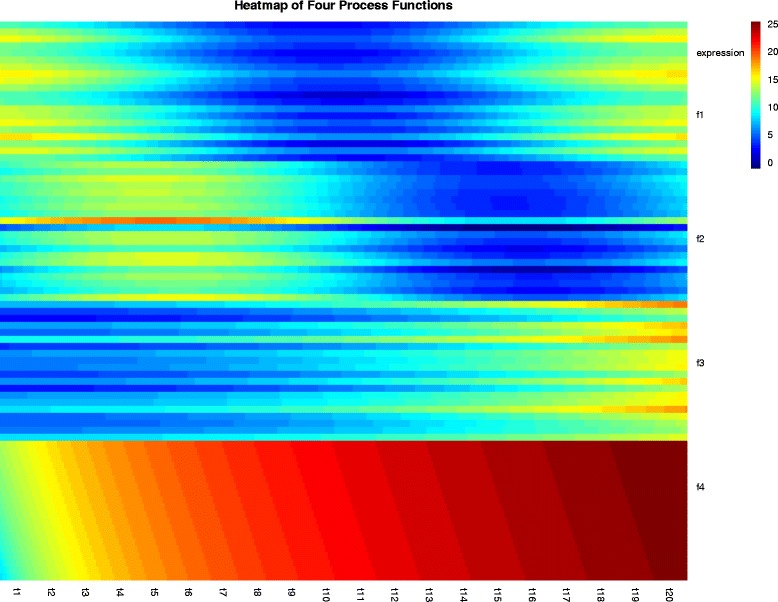
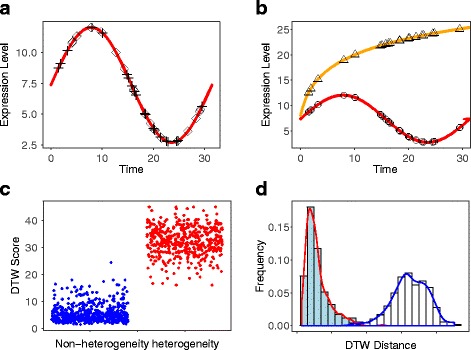
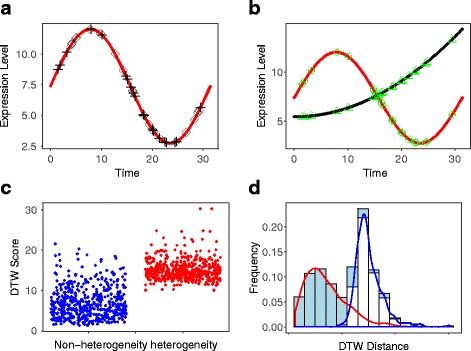
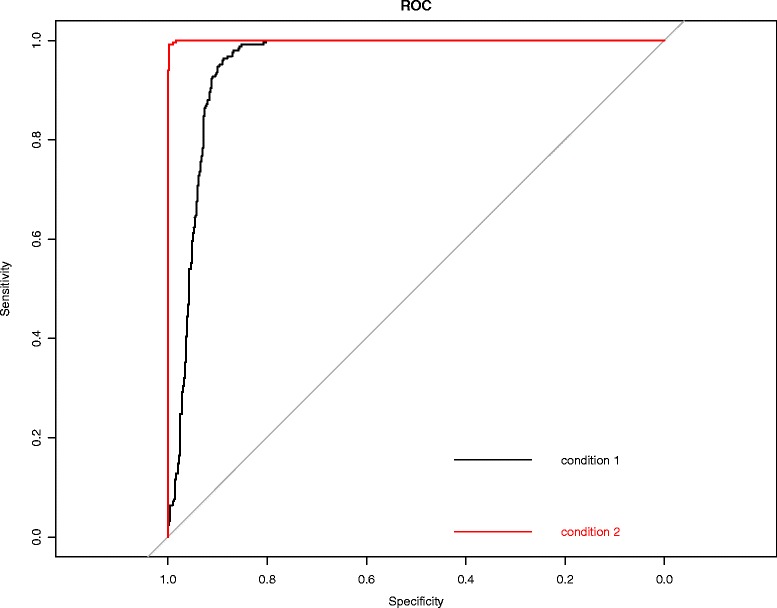
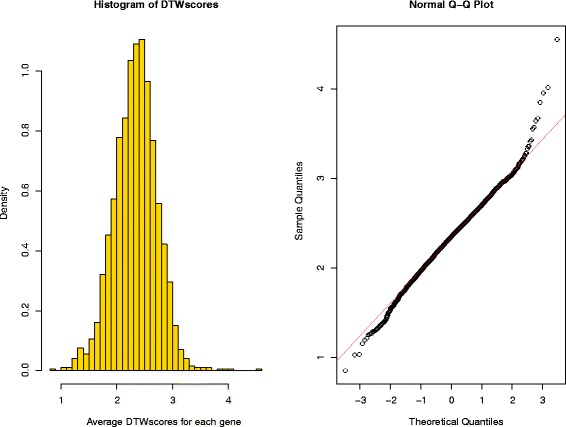
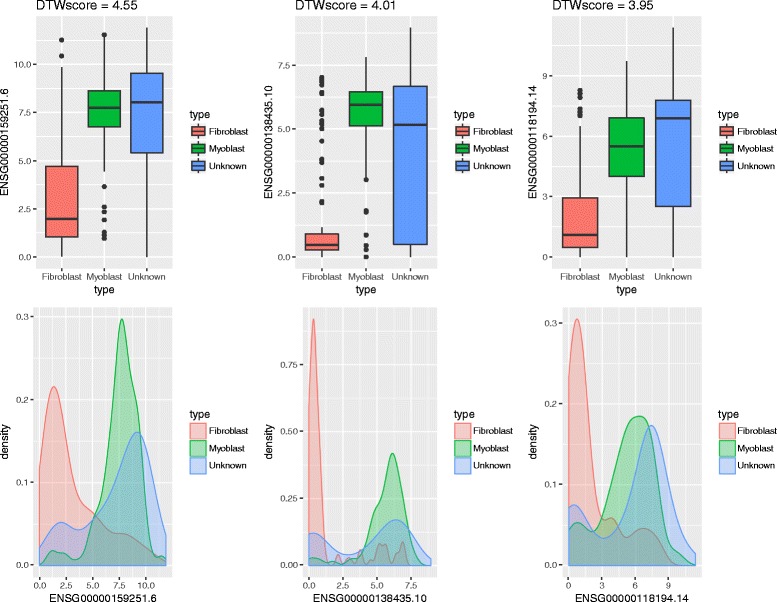
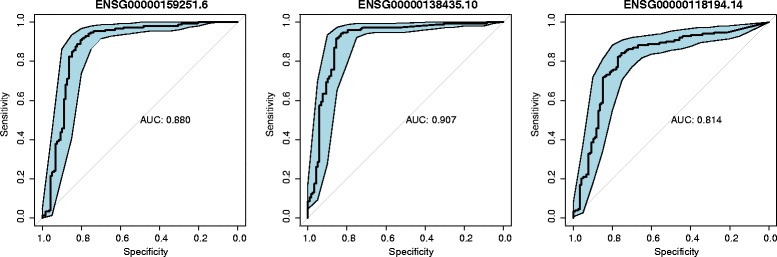
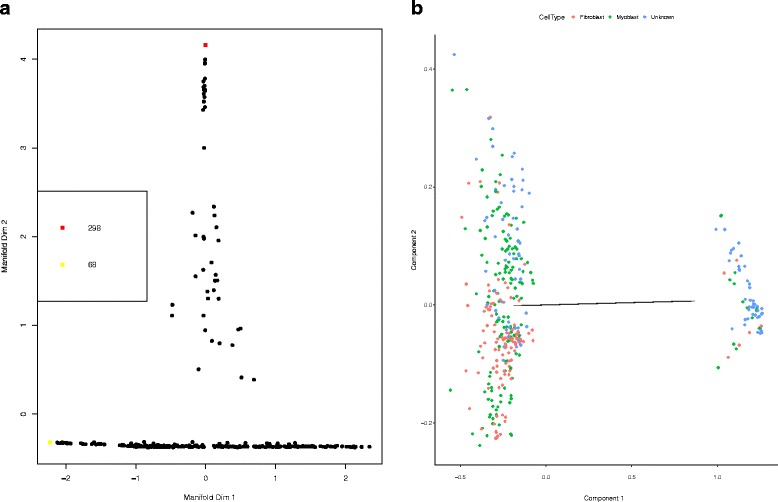
We present an algorithm based on the Dynamic Time Warping score (DTWscore) combined with time-series data, that enables the detection of gene expression changes across scRNA-seq samples and recovery of potential cell types from complex mixtures of multiple cell types.
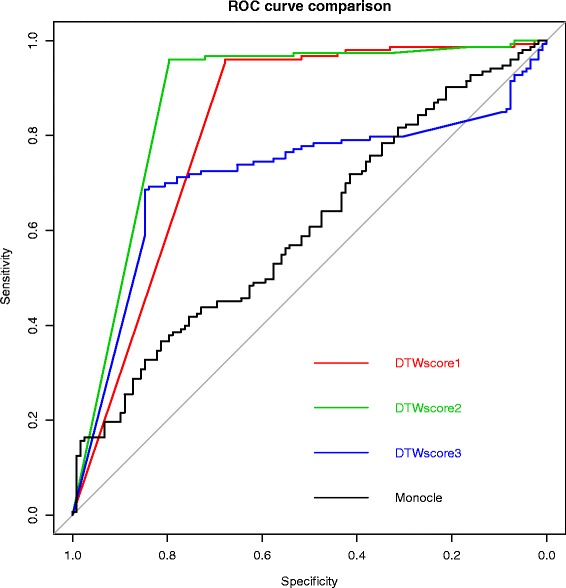
The DTWscore successfully classify cells of different types with the most highly variable genes from time-series scRNA-seq data. The study was confined to methods that are implemented and available within the R framework. Sample datasets and R packages are available at https://github.com/xiaoxiaoxier/DTWscore .
单细胞RNA测序的发展使得生物学领域有了重大发现,从剖析复杂组织的组成到识别某些特殊细胞环境中的新型细胞类型和动态变化。然而,在转录组分析中,在多个时间点收集大规模单细胞RNA测序(scRNA-seq)数据仍然是有效测量基因表达模式的一个挑战。
我们提出了一种基于动态时间规整分数(DTWscore)并结合时间序列数据的算法,该算法能够检测跨scRNA-seq样本的基因表达变化,并从多种细胞类型的复杂混合物中恢复潜在的细胞类型。
DTWscore成功地利用时间序列scRNA-seq数据中变化最大的基因对不同类型的细胞进行了分类。该研究局限于在R框架内实现且可用的方法。样本数据集和R包可在https://github.com/xiaoxiaoxier/DTWscore获取。