Khan Atif, Katanic Dejan, Thakar Juilee
Department of Microbiology and Immunology, University of Rochester, Rochester, NY, 14642, USA.
Department of Biostatistics and Computational Biology, University of Rochester, Rochester, NY, 14642, USA.
BMC Bioinformatics. 2017 Jun 6;18(1):295. doi: 10.1186/s12859-017-1669-x.
Despite advances in the gene-set enrichment analysis methods; inadequate definitions of gene-sets cause a major limitation in the discovery of novel biological processes from the transcriptomic datasets. Typically, gene-sets are obtained from publicly available pathway databases, which contain generalized definitions frequently derived by manual curation. Recently unsupervised clustering algorithms have been proposed to identify gene-sets from transcriptomics datasets deposited in public domain. These data-driven definitions of the gene-sets can be context-specific revealing novel biological mechanisms. However, the previously proposed algorithms for identification of data-driven gene-sets are based on hard clustering which do not allow overlap across clusters, a characteristic that is predominantly observed across biological pathways.
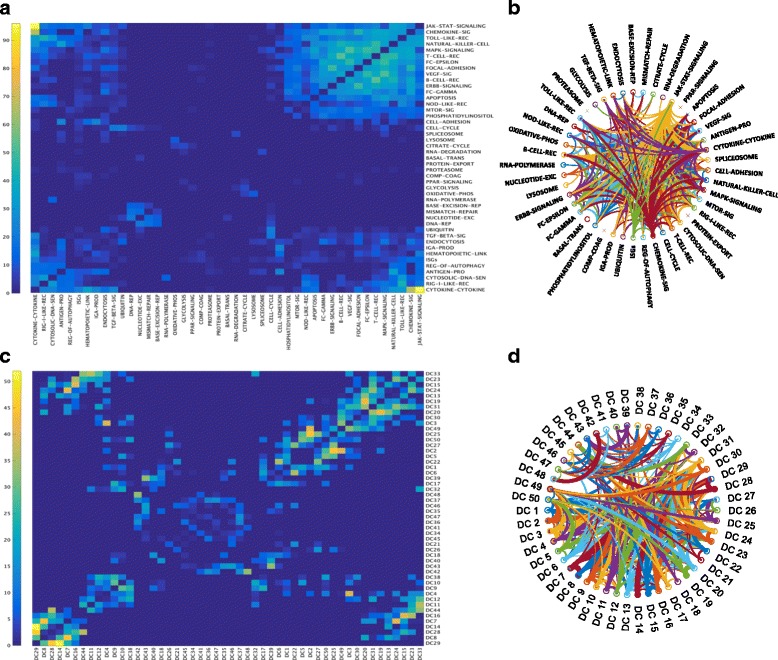
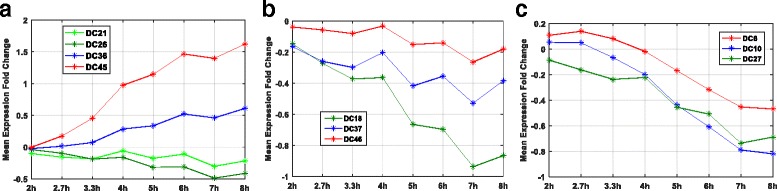
We developed a pipeline using fuzzy-C-means (FCM) soft clustering approach to identify gene-sets which recapitulates topological characteristics of biological pathways. Specifically, we apply our pipeline to derive gene-sets from transcriptomic data measuring response of monocyte derived dendritic cells and A549 epithelial cells to influenza infections. Our approach apply Ward's method for the selection of initial conditions, optimize parameters of FCM algorithm for human cell-specific transcriptomic data and identify robust gene-sets along with versatile viral responsive genes.
We validate our gene-sets and demonstrate that by identifying genes associated with multiple gene-sets, FCM clustering algorithm significantly improves interpretation of transcriptomic data facilitating investigation of novel biological processes by leveraging on transcriptomic data available in the public domain. We develop an interactive 'Fuzzy Inference of Gene-sets (FIGS)' package (GitHub: https://github.com/Thakar-Lab/FIGS ) to facilitate use of of pipeline. Future extension of FIGS across different immune cell-types will improve mechanistic investigation followed by high-throughput omics studies.
尽管基因集富集分析方法取得了进展,但基因集定义不充分仍是从转录组数据集中发现新生物过程的主要限制。通常,基因集是从公开可用的通路数据库中获取的,这些数据库包含通过人工整理频繁得出的广义定义。最近,有人提出了无监督聚类算法,用于从公共领域存储的转录组数据集中识别基因集。这些数据驱动的基因集定义可能是特定于上下文的,揭示了新的生物学机制。然而,先前提出的用于识别数据驱动基因集的算法基于硬聚类,不允许聚类之间存在重叠,而这一特征在生物通路中普遍存在。
我们开发了一种使用模糊C均值(FCM)软聚类方法的流程来识别基因集,该流程概括了生物通路的拓扑特征。具体而言,我们应用我们的流程从测量单核细胞衍生的树突状细胞和A549上皮细胞对流感感染反应的转录组数据中导出基因集。我们的方法应用沃德方法来选择初始条件,针对人类细胞特异性转录组数据优化FCM算法的参数,并识别出稳健的基因集以及通用的病毒反应基因。
我们验证了我们的基因集,并证明通过识别与多个基因集相关的基因,FCM聚类算法显著改善了对转录组数据的解释,通过利用公共领域中可用的转录组数据促进了对新生物过程的研究。我们开发了一个交互式的“基因集模糊推理(FIGS)”软件包(GitHub:https://github.com/Thakar-Lab/FIGS ),以方便使用该流程。FIGS在不同免疫细胞类型上的未来扩展将改善高通量组学研究后的机制研究。