Qiu Xing, Wu Shuang, Hilchey Shannon P, Thakar Juilee, Liu Zhi-Ping, Welle Stephen L, Henn Alicia D, Wu Hulin, Zand Martin S
Department of Biostatistics and Computational Biology, University of Rochester Medical Center, Rochester, NY, 14642, United States of America.
Department of Medicine, University of Rochester Medical Center, Rochester, NY, 14642, United States of America.
PLoS One. 2015 Sep 28;10(9):e0138110. doi: 10.1371/journal.pone.0138110. eCollection 2015.
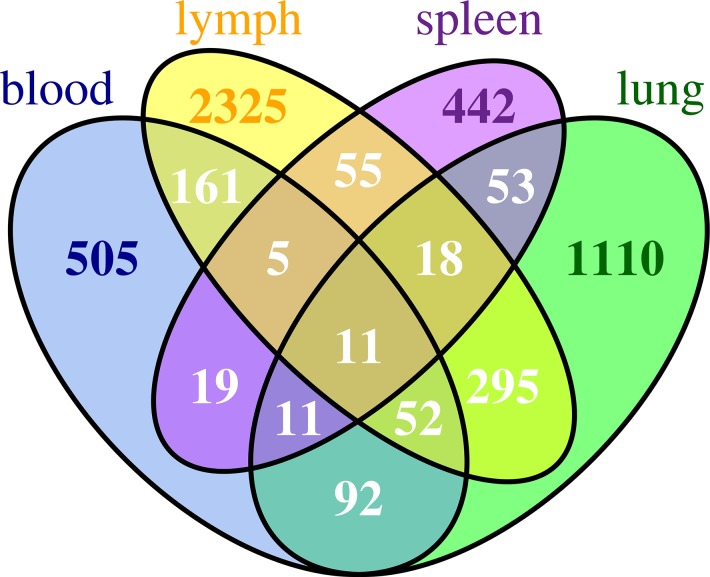
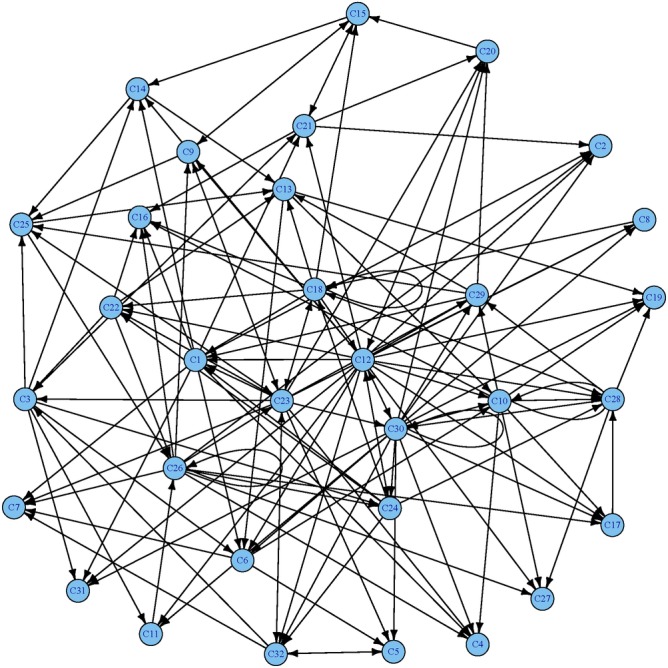
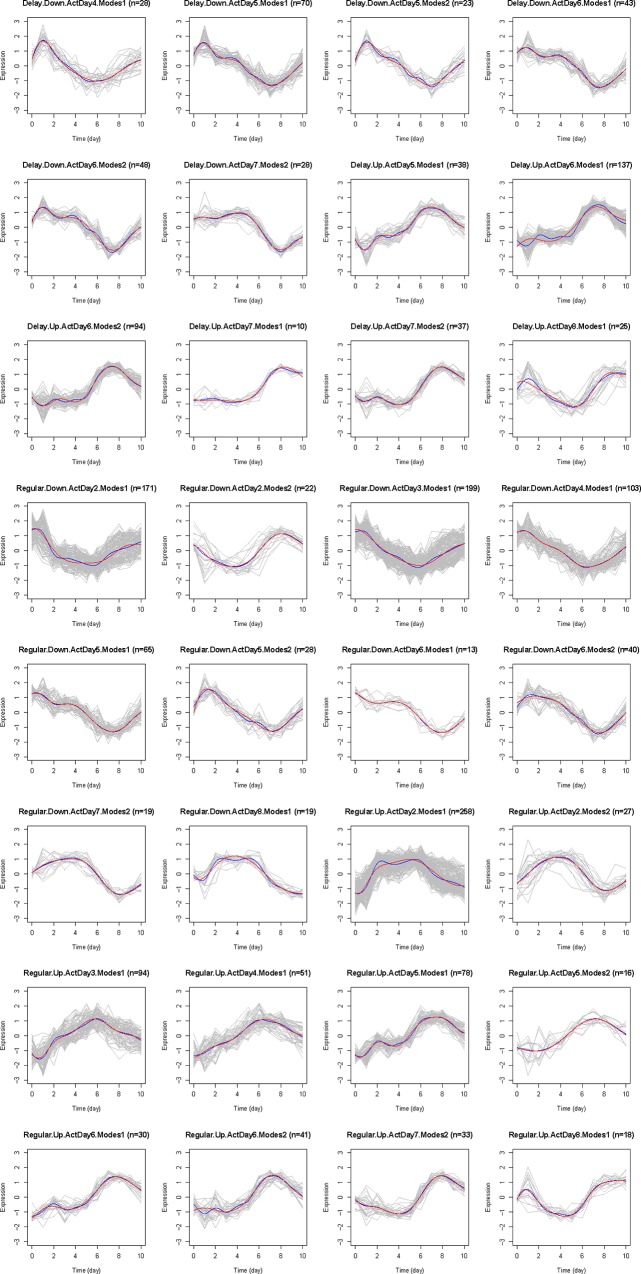
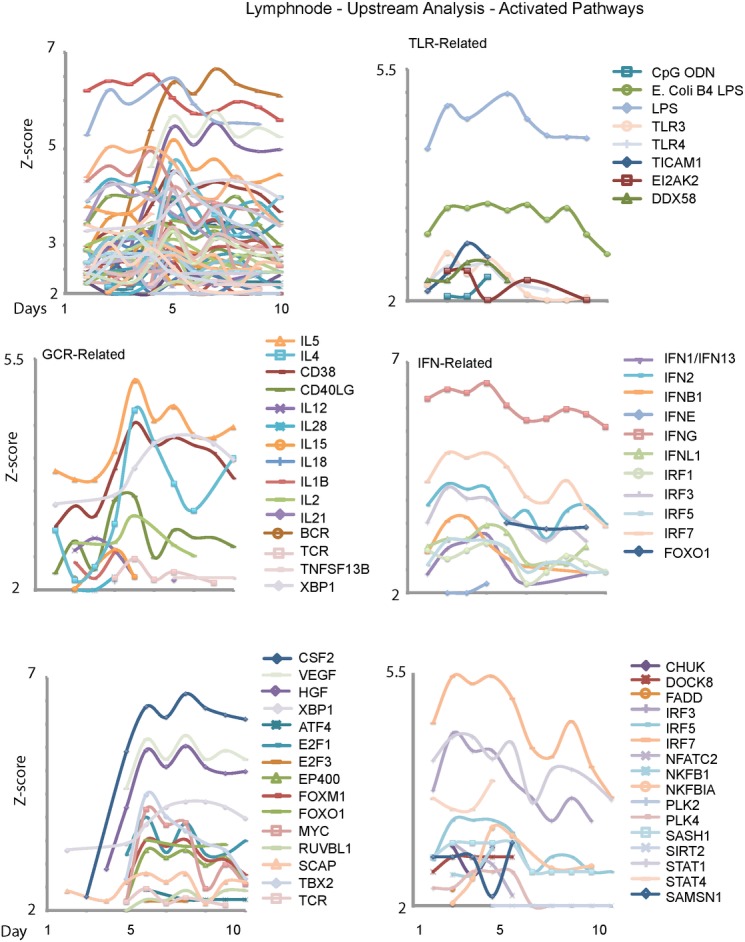
Current approaches to study transcriptional profiles post influenza infection typically rely on tissue sampling from one or two sites at a few time points, such as spleen and lung in murine models. In this study, we infected female C57/BL6 mice intranasally with mouse-adapted H3N2/Hong Kong/X31 avian influenza A virus, and then analyzed the gene expression profiles in four different compartments (blood, lung, mediastinal lymph nodes, and spleen) over 11 consecutive days post infection. These data were analyzed by an advanced statistical procedure based on ordinary differential equation (ODE) modeling. Vastly different lists of significant genes were identified by the same statistical procedure in each compartment. Only 11 of them are significant in all four compartments. We classified significant genes in each compartment into co-expressed modules based on temporal expression patterns. We then performed functional enrichment analysis on these co-expression modules and identified significant pathway and functional motifs. Finally, we used an ODE based model to reconstruct gene regulatory network (GRN) for each compartment and studied their network properties.
目前研究流感感染后转录谱的方法通常依赖于在几个时间点从一两个部位采集组织样本,比如在小鼠模型中的脾脏和肺。在本研究中,我们通过鼻内接种将适应小鼠的H3N2/香港/X31甲型禽流感病毒感染雌性C57/BL6小鼠,然后在感染后的连续11天分析四个不同腔室(血液、肺、纵隔淋巴结和脾脏)中的基因表达谱。这些数据通过基于常微分方程(ODE)建模的先进统计程序进行分析。在每个腔室中,通过相同的统计程序鉴定出了截然不同的显著基因列表。其中只有11个在所有四个腔室中都显著。我们根据时间表达模式将每个腔室中的显著基因分类为共表达模块。然后我们对这些共表达模块进行功能富集分析,并鉴定出显著的途径和功能基序。最后,我们使用基于ODE的模型为每个腔室重建基因调控网络(GRN)并研究其网络特性。