Sezione di Biomedicina e Endocrinologia, Dipartimento di Scienze Farmacologiche e Biomolecolari (DiSFeB), Centro di Eccellenza sulle Malattie Neurodegenerative, Università degli Studi di Milano, Via Balzaretti 9, 20133, Milan, Italy.
C. Mondino National Neurological Institute, Pavia, Italy.
Cell Stress Chaperones. 2018 Jan;23(1):1-12. doi: 10.1007/s12192-017-0806-9. Epub 2017 Jun 12.
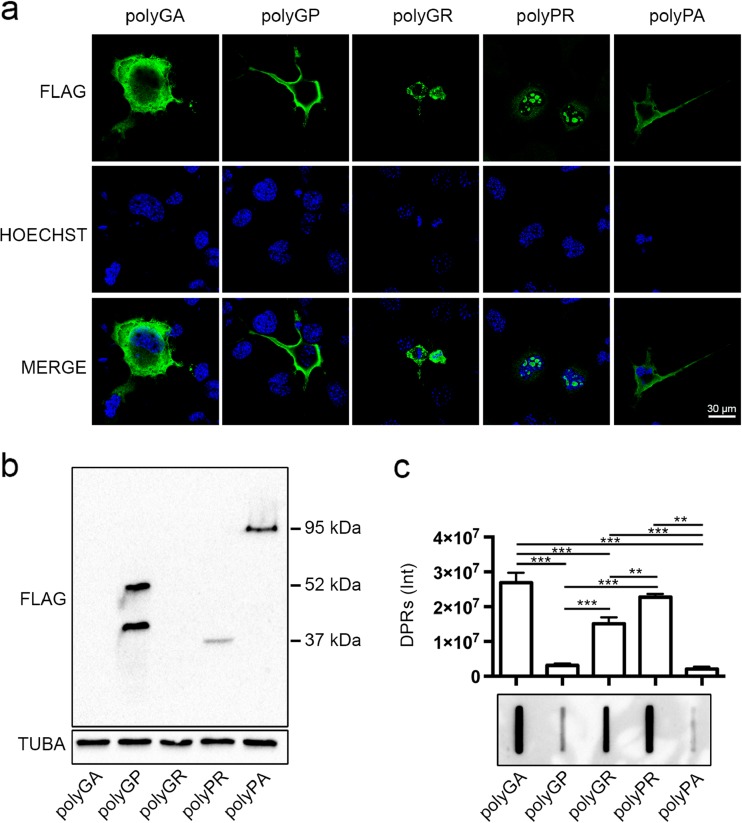
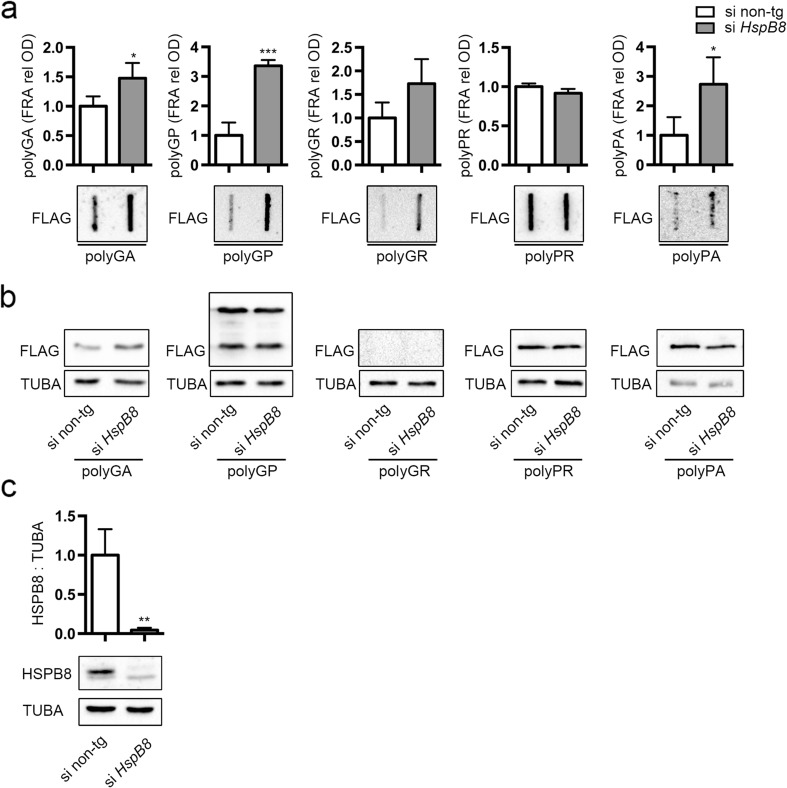
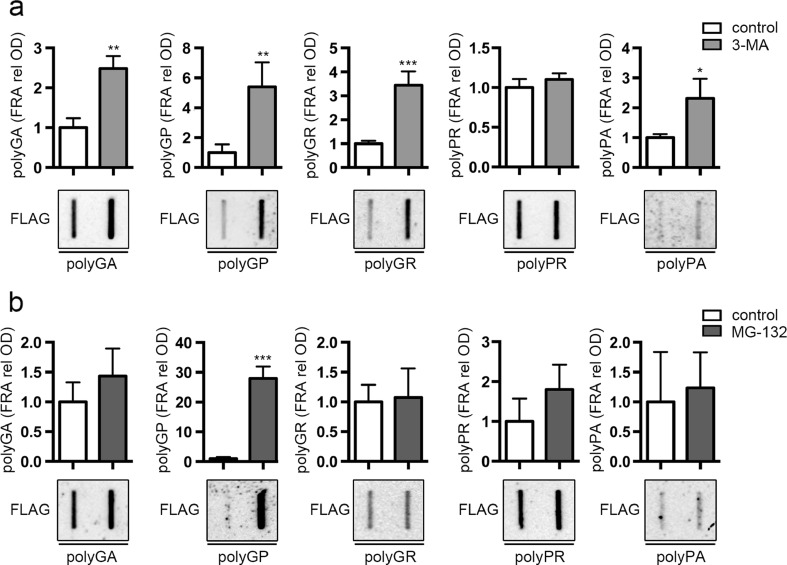
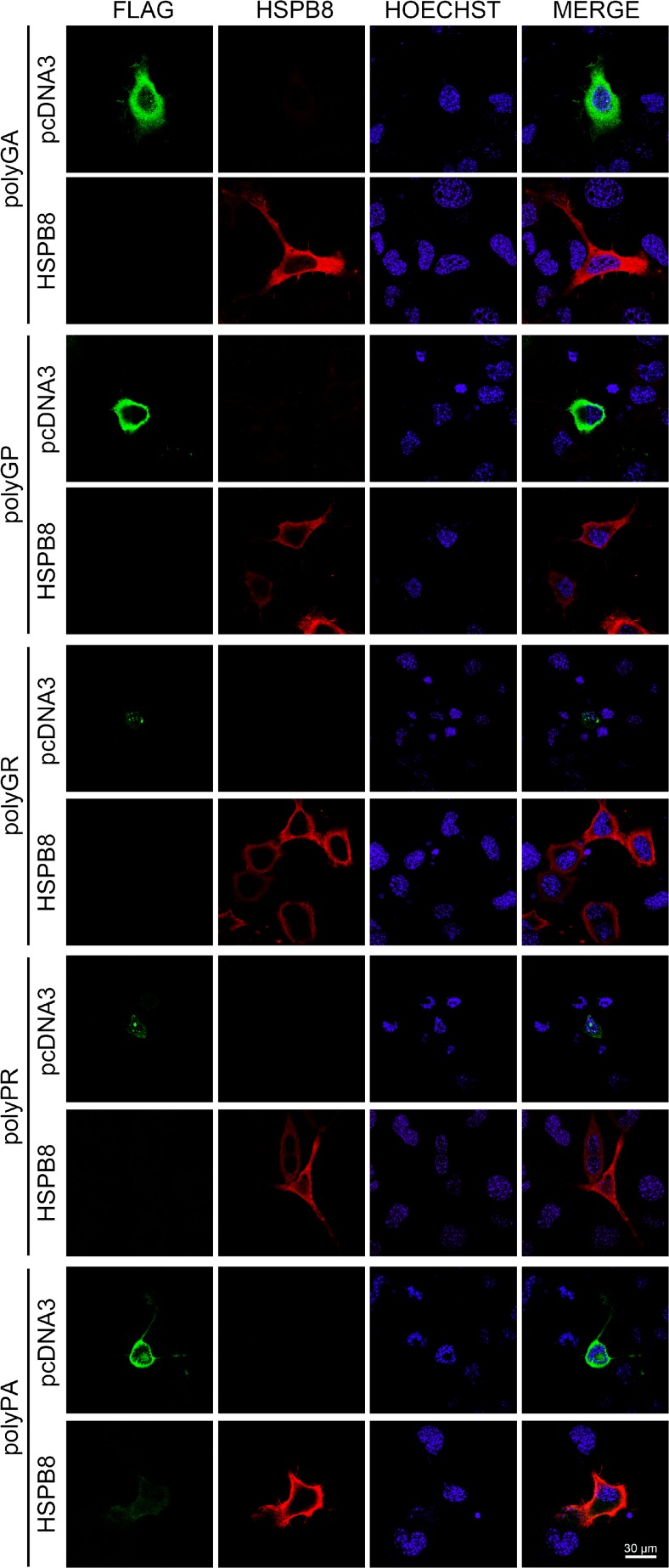
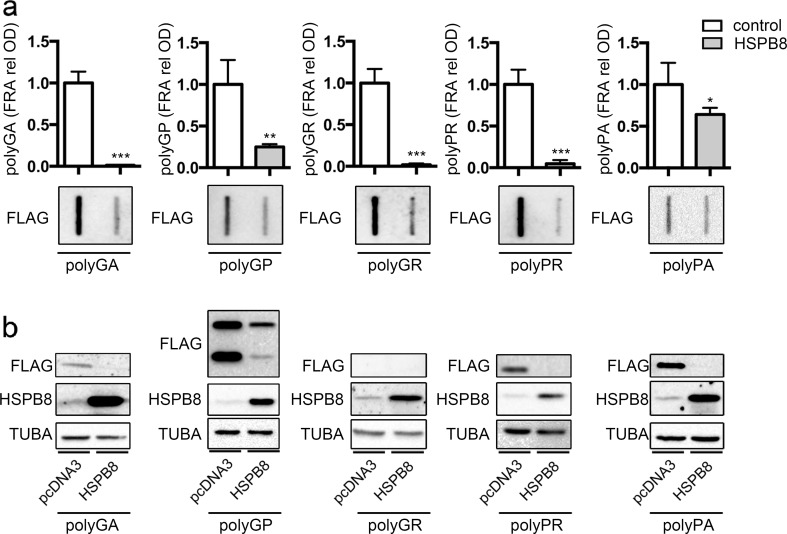
Amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) are two neurodegenerative diseases in which similar pathogenic mechanisms are involved. Both diseases associate to the high propensity of specific misfolded proteins, like TDP-43 or FUS, to mislocalize and aggregate. This is partly due to their intrinsic biophysical properties and partly as a consequence of failure of the neuronal protein quality control (PQC) system. Several familial ALS/FTD cases are linked to an expansion of a repeated G4C2 hexanucleotide sequence present in the C9ORF72 gene. The G4C2, which localizes in an untranslated region of the C9ORF72 transcript, drives an unconventional repeat-associated ATG-independent translation. This leads to the synthesis of five different dipeptide repeat proteins (DPRs), which are not "classical" misfolded proteins, but generate aberrant aggregation-prone unfolded conformations poorly removed by the PQC system. The DPRs accumulate into p62/SQSTM1 and ubiquitin positive inclusions. Here, we analyzed the biochemical behavior of the five DPRs in immortalized motoneurons. Our data suggest that while the DPRs are mainly processed via autophagy, this system is unable to fully clear their aggregated forms, and thus they tend to accumulate in basal conditions. Overexpression of the small heat shock protein B8 (HSPB8), which facilitates the autophagy-mediated disposal of a large variety of classical misfolded aggregation-prone proteins, significantly decreased the accumulation of most DPR insoluble species. Thus, the induction of HSPB8 might represent a valid approach to decrease DPR-mediated toxicity and maintain motoneuron viability.
肌萎缩侧索硬化症(ALS)和额颞叶痴呆(FTD)是两种神经退行性疾病,其中涉及相似的致病机制。这两种疾病都与特定错误折叠蛋白(如 TDP-43 或 FUS)易位和聚集的高倾向相关。这部分是由于它们的固有生物物理特性,部分是由于神经元蛋白质质量控制(PQC)系统失效。几种家族性 ALS/FTD 病例与 C9ORF72 基因中存在的 G4C2 六核苷酸重复序列的扩展有关。G4C2 定位于 C9ORF72 转录本的非翻译区,驱动非常规的重复相关 ATG 非依赖性翻译。这导致合成五种不同的二肽重复蛋白(DPR),它们不是“经典”错误折叠蛋白,但产生异常聚集倾向的未折叠构象,无法被 PQC 系统有效清除。DPR 积累到 p62/SQSTM1 和泛素阳性包涵体中。在这里,我们分析了五种 DPR 在永生化运动神经元中的生化行为。我们的数据表明,尽管 DPR 主要通过自噬途径进行处理,但该系统无法完全清除其聚集形式,因此它们在基础条件下容易积累。小分子热休克蛋白 B8(HSPB8)的过表达促进了多种经典错误折叠聚集倾向蛋白的自噬介导处理,显著降低了大多数 DPR 不溶性物质的积累。因此,诱导 HSPB8 可能是降低 DPR 介导的毒性并维持运动神经元活力的有效方法。