Zhang Kuixing, Lu Yuxin, Harley Kevin T, Tran Minh-Ha
Department of Pathology and Laboratory Medicine, University of California, Orange, CA, USA.
Department of Internal Medicine, Division of Nephrology and Hypertension, Irvine School of Medicine, University of California, Orange, CA, USA.
Hematol Rep. 2017 Jun 1;9(2):7053. doi: 10.4081/hr.2017.7053.
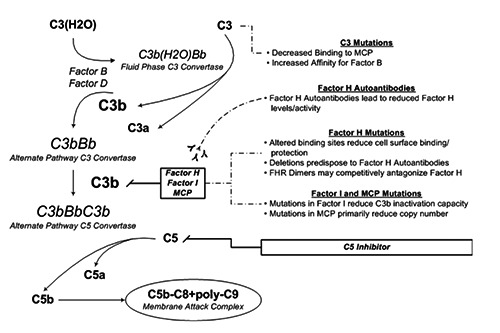
Atypical hemolytic uremic syndrome (aHUS) is a disease characterized by the triad of microangiopathic hemolytic anemia, thrombocytopenia and acute kidney injury. The histopathologic lesions of aHUS include thrombotic microangiopathy involving the glomerular capillaries and thrombosis involving arterioles or interlobar arteries. Extra-renal manifestations occur in up to 20% of patients. The majority of aHUS is caused by complement system defects impairing ordinary regulatory mechanisms. Activating events therefore lead to unbridled, ongoing complement activity producing widespread endothelial injury. Pathologic mutations include those resulting in loss-of-function in a complement regulatory gene ( or ) or gain-of-function in an effector gene ( or ). Treatment with the late complement inhibitor, eculizumab - a monoclonal antibody directed against C5 - is effective.
非典型溶血性尿毒症综合征(aHUS)是一种以微血管病性溶血性贫血、血小板减少和急性肾损伤三联征为特征的疾病。aHUS的组织病理学病变包括累及肾小球毛细血管的血栓性微血管病以及累及小动脉或叶间动脉的血栓形成。高达20%的患者会出现肾外表现。大多数aHUS是由损害正常调节机制的补体系统缺陷引起的。因此,激活事件会导致不受控制的、持续的补体活性,从而产生广泛的内皮损伤。病理突变包括导致补体调节基因功能丧失(或)或效应基因功能获得(或)的突变。使用晚期补体抑制剂依库珠单抗(一种针对C5的单克隆抗体)进行治疗是有效的。