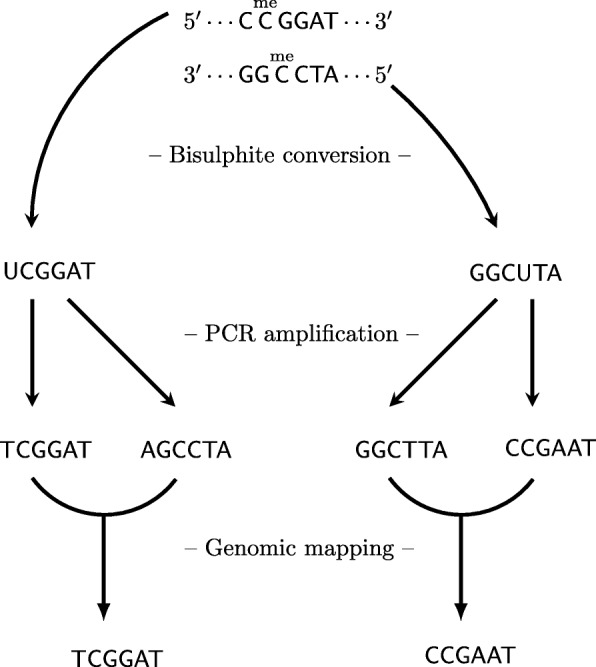
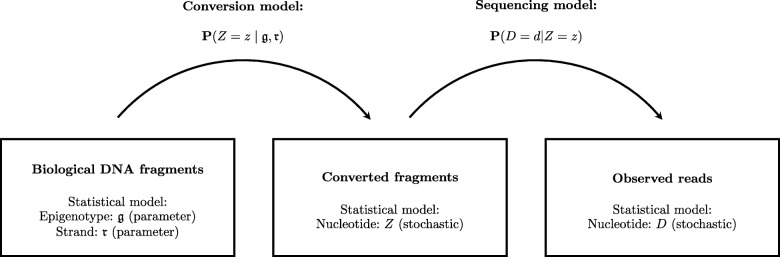
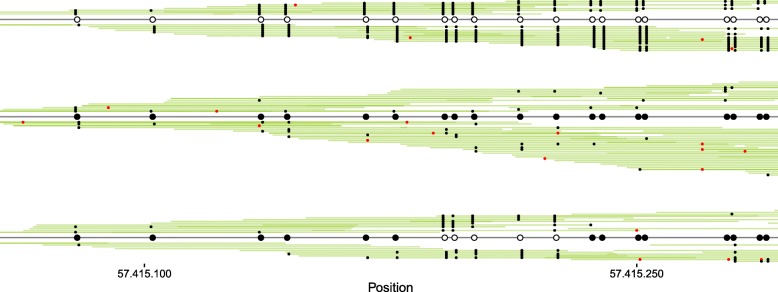
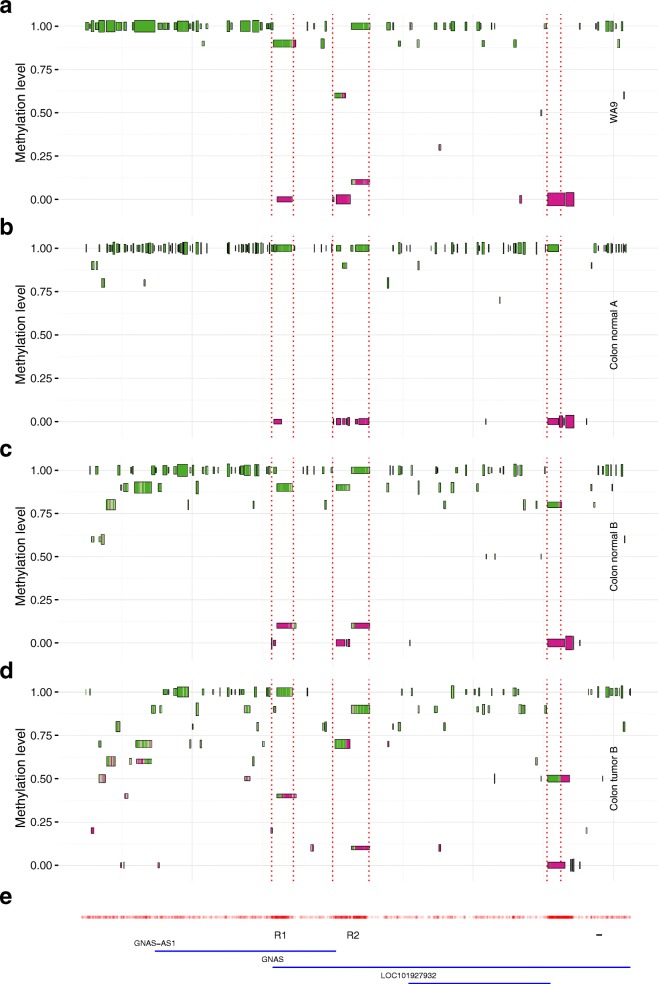
Vincent Martin, Mundbjerg Kamilla, Skou Pedersen Jakob, Liang Gangning, Jones Peter A, Ørntoft Torben Falck, Dalsgaard Sørensen Karina, Wiuf Carsten
Department of Mathematical Sciences, University of Copenhagen, Copenhagen, 2100, Denmark.
USC Norris Comprehensive Cancer Center, Keck School of Medicine, Los Angeles, 90089-9176, CA, USA.
Genome Biol. 2017 Feb 21;18(1):38. doi: 10.1186/s13059-017-1168-4.
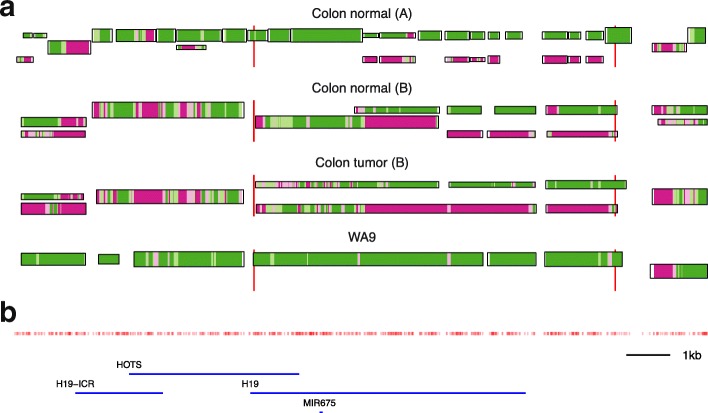
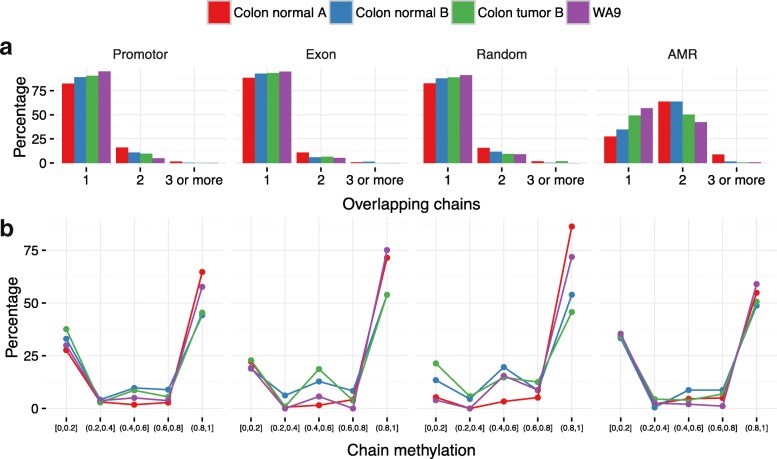
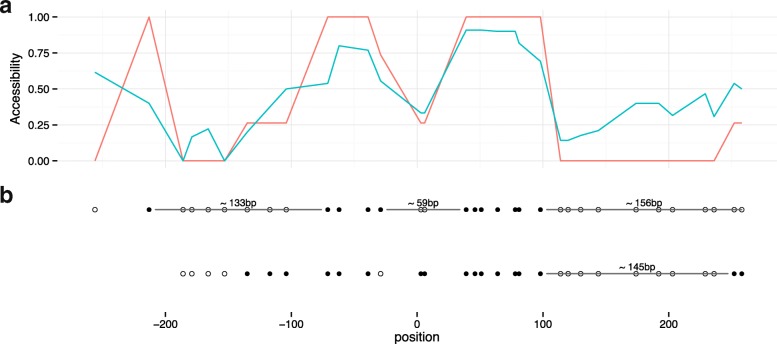
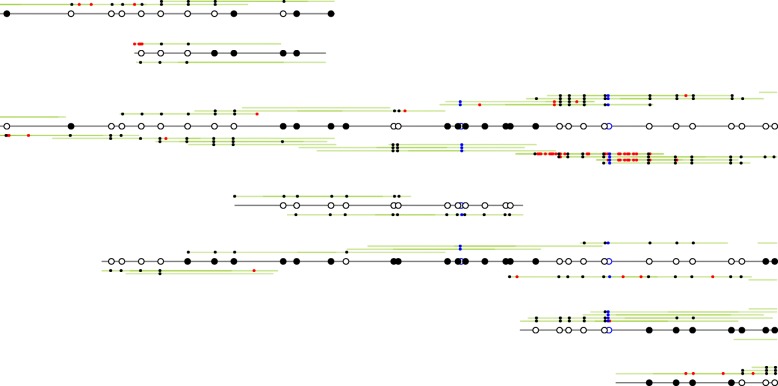
The study of epigenetic heterogeneity at the level of individual cells and in whole populations is the key to understanding cellular differentiation, organismal development, and the evolution of cancer. We develop a statistical method, epiG, to infer and differentiate between different epi-allelic haplotypes, annotated with CpG methylation status and DNA polymorphisms, from whole-genome bisulfite sequencing data, and nucleosome occupancy from NOMe-seq data. We demonstrate the capabilities of the method by inferring allele-specific methylation and nucleosome occupancy in cell lines, and colon and tumor samples, and by benchmarking the method against independent experimental data.
在单个细胞水平和整个群体中研究表观遗传异质性是理解细胞分化、机体发育和癌症演变的关键。我们开发了一种统计方法epiG,用于从全基因组亚硫酸氢盐测序数据中推断并区分不同的表观等位基因单倍型(标注有CpG甲基化状态和DNA多态性),以及从NOMe-seq数据中推断核小体占有率。我们通过推断细胞系、结肠和肿瘤样本中的等位基因特异性甲基化和核小体占有率,并将该方法与独立实验数据进行基准测试,来证明该方法的能力。