Mudge J F, Martyniuk C J, Houlahan J E
Department of Biology, Canadian Rivers Institute, University of New Brunswick, Saint John, NB, E2L 4L5, Canada.
Center for Environmental and Human Toxicology & Department of Physiological Sciences, UF Genetics Institute, University of Florida, Gainesville, Florida, 32611, USA.
BMC Bioinformatics. 2017 Jun 21;18(1):312. doi: 10.1186/s12859-017-1728-3.
Transcriptomic approaches (microarray and RNA-seq) have been a tremendous advance for molecular science in all disciplines, but they have made interpretation of hypothesis testing more difficult because of the large number of comparisons that are done within an experiment. The result has been a proliferation of techniques aimed at solving the multiple comparisons problem, techniques that have focused primarily on minimizing Type I error with little or no concern about concomitant increases in Type II errors. We have previously proposed a novel approach for setting statistical thresholds with applications for high throughput omics-data, optimal α, which minimizes the probability of making either error (i.e. Type I or II) and eliminates the need for post-hoc adjustments.
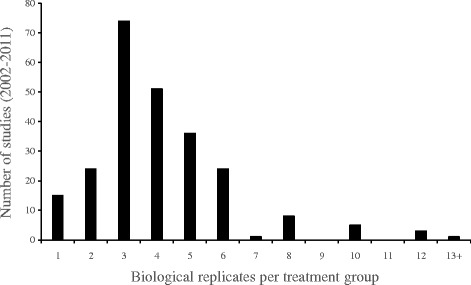
A meta-analysis of 242 microarray studies extracted from the peer-reviewed literature found that current practices for setting statistical thresholds led to very high Type II error rates. Further, we demonstrate that applying the optimal α approach results in error rates as low or lower than error rates obtained when using (i) no post-hoc adjustment, (ii) a Bonferroni adjustment and (iii) a false discovery rate (FDR) adjustment which is widely used in transcriptome studies.
We conclude that optimal α can reduce error rates associated with transcripts in both microarray and RNA-seq experiments, but point out that improved statistical techniques alone cannot solve the problems associated with high throughput datasets - these approaches need to be coupled with improved experimental design that considers larger sample sizes and/or greater study replication.
转录组学方法(微阵列和RNA测序)在所有学科的分子科学领域都取得了巨大进展,但由于在一个实验中要进行大量比较,使得假设检验的解释变得更加困难。结果是大量旨在解决多重比较问题的技术不断涌现,这些技术主要侧重于将I型错误最小化,而很少或根本不考虑随之而来的II型错误增加。我们之前提出了一种用于设置统计阈值的新方法——最优α,适用于高通量组学数据,该方法能将犯任何一种错误(即I型或II型)的概率最小化,并且无需进行事后调整。
对从同行评审文献中提取的242项微阵列研究进行的荟萃分析发现,当前设置统计阈值的做法导致了非常高的II型错误率。此外,我们证明,应用最优α方法所得到的错误率与使用(i)不进行事后调整、(ii)Bonferroni调整和(iii)转录组研究中广泛使用的错误发现率(FDR)调整时所获得的错误率一样低或更低。
我们得出结论,最优α可以降低微阵列和RNA测序实验中转录本相关的错误率,但指出仅靠改进的统计技术无法解决与高通量数据集相关的问题——这些方法需要与考虑更大样本量和/或更高研究重复性的改进实验设计相结合。