Lee Paul R, Johnson Tory P, Gnanapavan Sharmilee, Giovannoni Gavin, Wang Tongguang, Steiner Joseph P, Medynets Marie, Vaal Mark J, Gartner Valerie, Nath Avindra
Section of Infections of the Nervous System, National Institute of Neurological Disorders and Stroke, National Institutes of Health, 10 Center Drive, Building 10, Room CRC 3-2563, Bethesda, MD, 20892, USA.
Centre for Neuroscience and Trauma, Blizard Institute, Barts and The London School of Medicine and Dentistry, London, UK.
J Neuroinflammation. 2017 Jun 27;14(1):131. doi: 10.1186/s12974-017-0901-y.
The cause of neurodegeneration in progressive forms of multiple sclerosis is unknown. We investigated the impact of specific neuroinflammatory markers on human neurons to identify potential therapeutic targets for neuroprotection against chronic inflammation.
Surface immunocytochemistry directly visualized protease-activated receptor-1 (PAR1) and interleukin-1 (IL-1) receptors on neurons in human postmortem cortex in patients with and without neuroinflammatory lesions. Viability of cultured neurons was determined after exposure to cerebrospinal fluid from patients with progressive multiple sclerosis or purified granzyme B and IL-1β. Inhibitors of PAR1 activation and of PAR1-associated second messenger signaling were used to elucidate a mechanism of neurotoxicity.
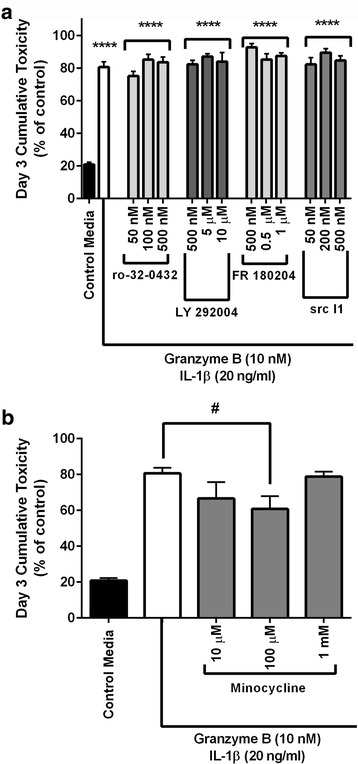
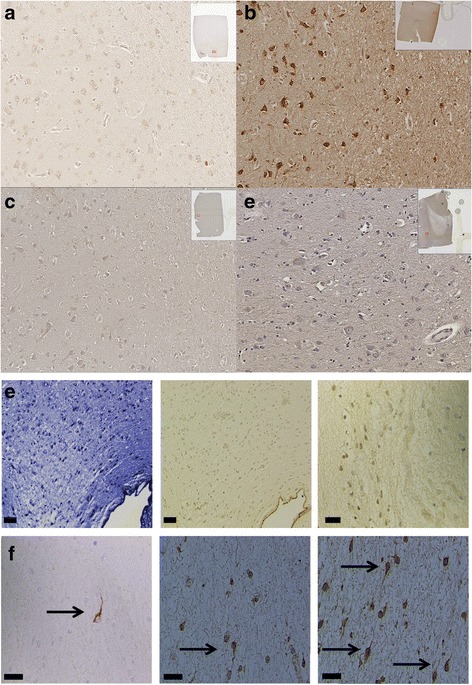
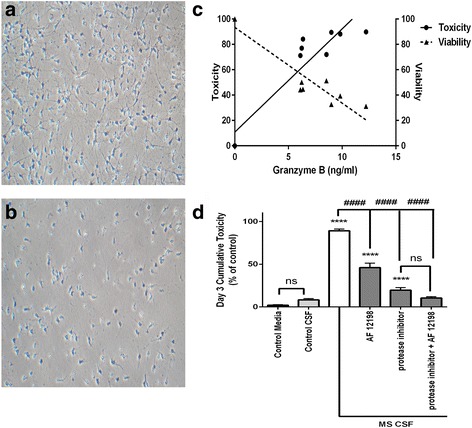
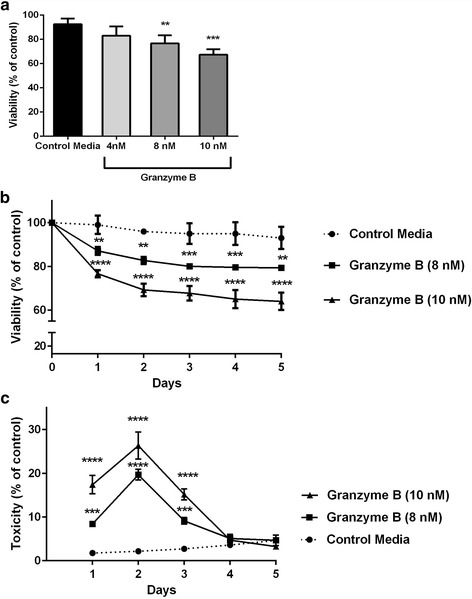
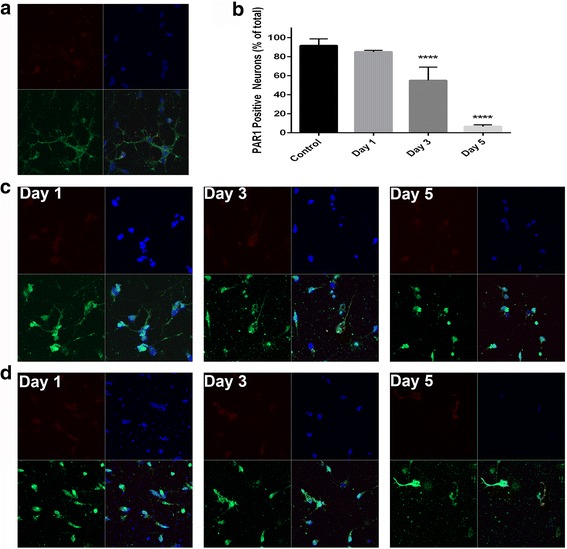
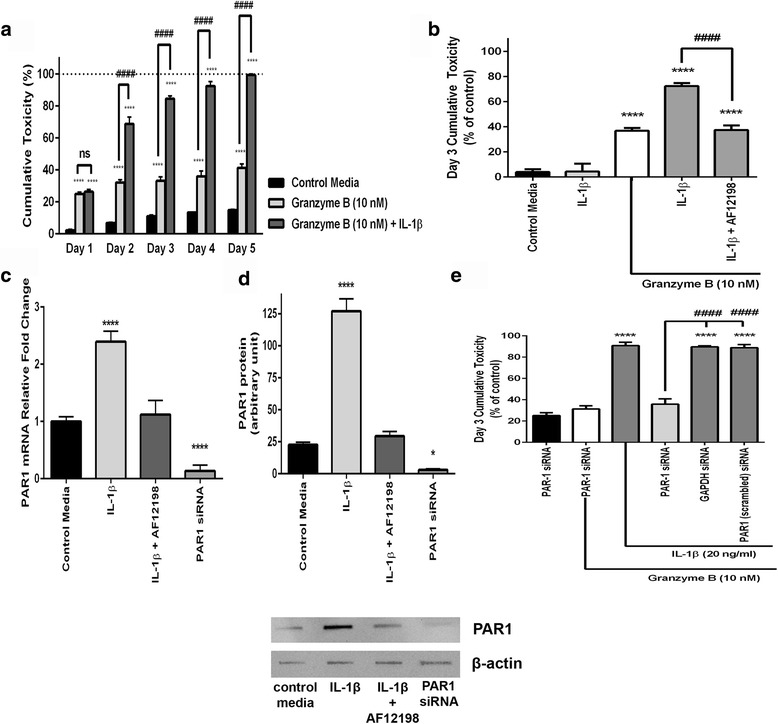
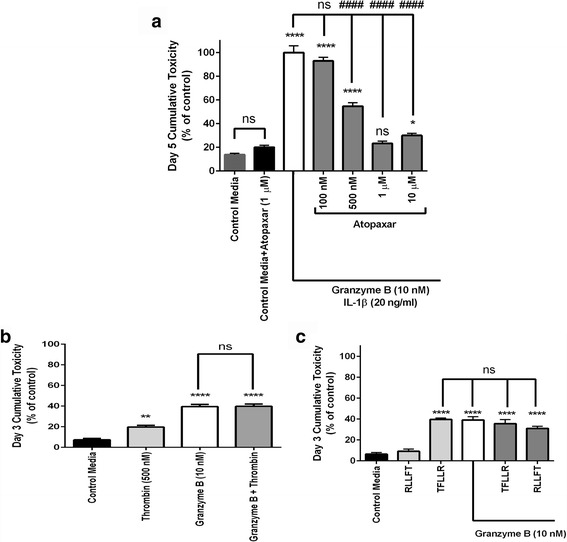
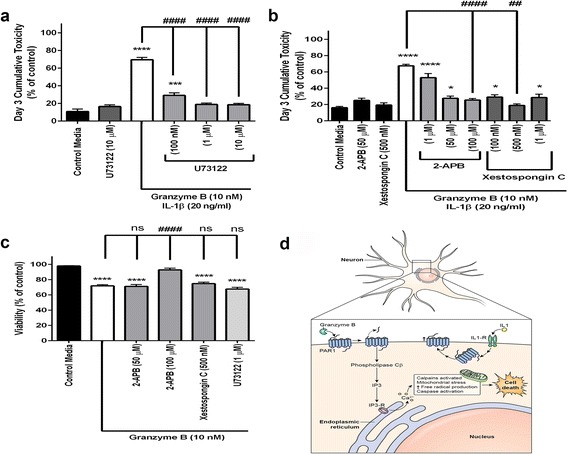
Immunohistochemistry of human post-mortem brain tissue demonstrated cells expressing higher amounts of PAR1 near and within subcortical lesions in patients with multiple sclerosis compared to control tissue. Human cerebrospinal fluid samples containing granzyme B and IL-1β were toxic to human neuronal cultures. Granzyme B was neurotoxic through activation of PAR1 and subsequently the phospholipase Cβ-IP3 second messenger system. Inhibition of PAR1 or IP3 prevented granzyme B toxicity. IL-1β enhanced granzyme B-mediated neurotoxicity by increasing PAR1 expression.
Neurons within the inflamed central nervous system are imperiled because they express more PAR1 and are exposed to a neurotoxic combination of both granzyme B and IL-1β. The effects of these inflammatory mediators may be a contributing factor in the progressive brain atrophy associated with neuroinflammatory diseases. Knowledge of how exposure to IL-1β and granzyme B act synergistically to cause neuronal death yields potential novel neuroprotective treatments for neuroinflammatory diseases.
多发性硬化症进展型神经退行性变的病因尚不清楚。我们研究了特定神经炎症标志物对人类神经元的影响,以确定针对慢性炎症的神经保护潜在治疗靶点。
通过表面免疫细胞化学直接观察有或无神经炎症病变的人类尸检皮质中神经元上的蛋白酶激活受体-1(PAR1)和白细胞介素-1(IL-1)受体。将培养的神经元暴露于进展型多发性硬化症患者的脑脊液、纯化的颗粒酶B和IL-1β后,测定其活力。使用PAR1激活抑制剂和PAR1相关的第二信使信号抑制剂来阐明神经毒性机制。
与对照组织相比,多发性硬化症患者尸检脑组织的免疫组织化学显示,皮质下病变附近和内部表达PAR1量较高的细胞。含有颗粒酶B和IL-1β的人类脑脊液样本对人类神经元培养物有毒性。颗粒酶B通过激活PAR1以及随后的磷脂酶Cβ-IP3第二信使系统具有神经毒性。抑制PAR1或IP3可预防颗粒酶B的毒性。IL-1β通过增加PAR1表达增强颗粒酶B介导的神经毒性。
炎症中枢神经系统内的神经元受到威胁,因为它们表达更多的PAR1,并暴露于颗粒酶B和IL-1β的神经毒性组合中。这些炎症介质的作用可能是与神经炎症性疾病相关的进行性脑萎缩的一个促成因素。了解暴露于IL-1β和颗粒酶B如何协同作用导致神经元死亡,为神经炎症性疾病带来了潜在的新型神经保护治疗方法。