Rorato Rodrigo, Borges Beatriz de Carvalho, Uchoa Ernane Torres, Antunes-Rodrigues José, Elias Carol Fuzeti, Elias Lucila Leico Kagohara
Department of Physiology, Ribeirao Preto Medical School, University of Sao Paulo, Sao Paulo 14049-900, Brazil.
Department of Molecular and Integrative Physiology, University of Michigan, Ann Arbor, MI 48109-5622, USA.
Int J Mol Sci. 2017 Jul 4;18(7):1431. doi: 10.3390/ijms18071431.
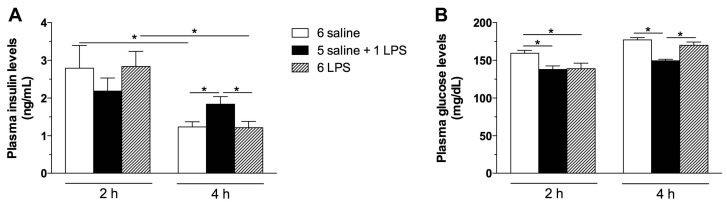
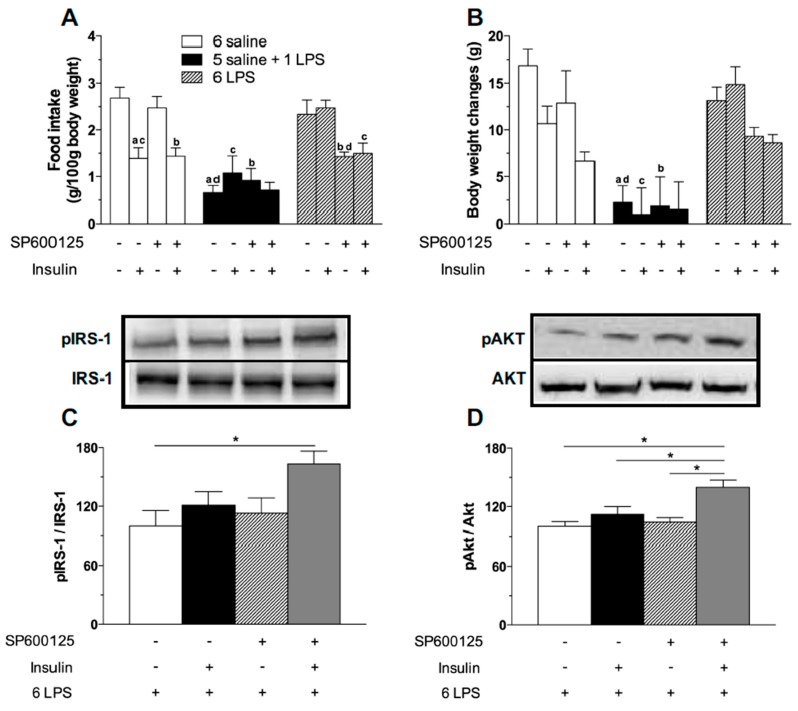
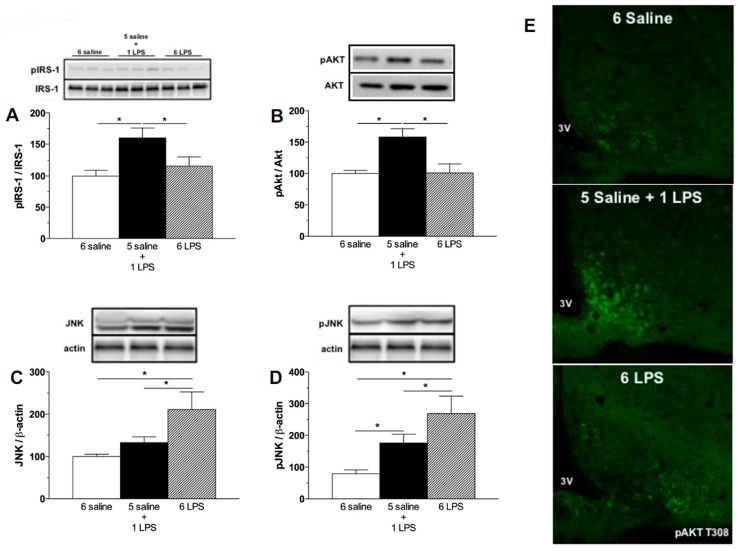
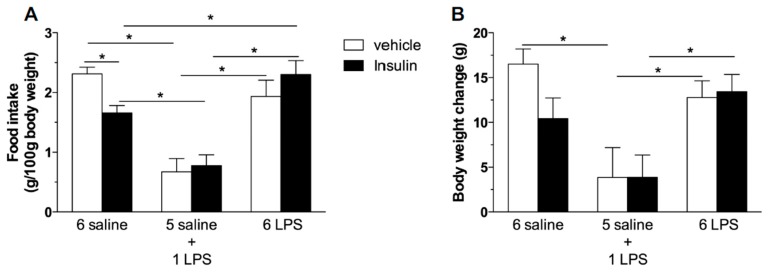
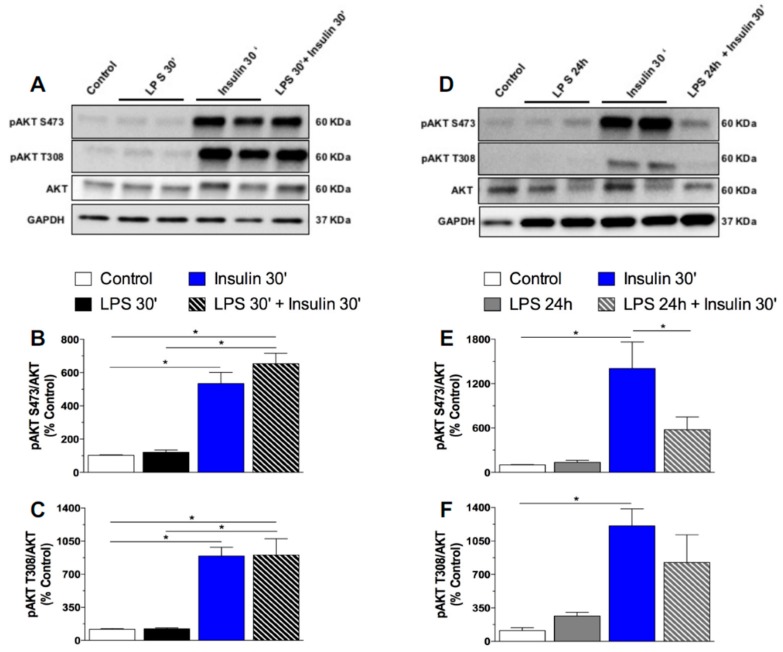
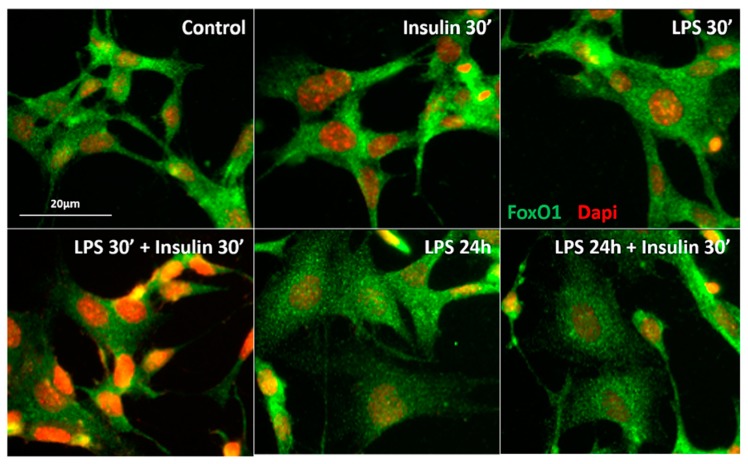
Metabolic endotoxemia contributes to low-grade inflammation in obesity, which causes insulin resistance due to the activation of intracellular proinflammatory pathways, such as the c-Jun N-terminal Kinase (JNK) cascade in the hypothalamus and other tissues. However, it remains unclear whether the proinflammatory process precedes insulin resistance or it appears because of the development of obesity. Hypothalamic low-grade inflammation was induced by prolonged lipopolysaccharide (LPS) exposure to investigate if central insulin resistance is induced by an inflammatory stimulus regardless of obesity. Male Wistar rats were treated with single (1 LPS) or repeated injections (6 LPS) of LPS (100 μg/kg, IP) to evaluate the phosphorylation of the insulin receptor substrate-1 (IRS1), Protein kinase B (AKT), and JNK in the hypothalamus. Single LPS increased the expression of pIRS1, pAKT, and pJNK, whereas the repeated LPS treatment failed to recruit pIRS1 and pAKT. The 6 LPS treated rats showed increased total JNK and pJNK. The 6 LPS rats became unresponsive to the hypophagic effect induced by central insulin administration (12 μM/5 μL, ICV). Prolonged exposure to LPS (24 h) impaired the insulin-induced AKT phosphorylation and the translocation of the transcription factor forkhead box protein O1 (FoxO1) from the nucleus to the cytoplasm of the cultured hypothalamic GT1-7 cells. Central administration of the JNK inhibitor (20 μM/5 μL, ICV) restored the ability of insulin to phosphorylate IRS1 and AKT in 6 LPS rats. The present data suggest that an increased JNK activity in the hypothalamus underlies the development of insulin resistance during prolonged exposure to endotoxins. Our study reveals that weight gain is not mandatory for the development of hypothalamic insulin resistance and the blockade of proinflammatory pathways could be useful for restoring the insulin signaling during prolonged low-grade inflammation as seen in obesity.
代谢性内毒素血症导致肥胖中的低度炎症,由于细胞内促炎途径(如下丘脑和其他组织中的c-Jun氨基末端激酶(JNK)级联反应)的激活,进而引起胰岛素抵抗。然而,促炎过程是先于胰岛素抵抗出现,还是由于肥胖的发展而出现,目前尚不清楚。通过长期暴露于脂多糖(LPS)诱导下丘脑低度炎症,以研究无论肥胖与否,炎症刺激是否会诱导中枢性胰岛素抵抗。对雄性Wistar大鼠进行单次(1次LPS)或重复注射(6次LPS)LPS(100μg/kg,腹腔注射),以评估下丘脑胰岛素受体底物-1(IRS1)、蛋白激酶B(AKT)和JNK的磷酸化情况。单次LPS增加了pIRS1、pAKT和pJNK的表达,而重复LPS处理未能募集pIRS1和pAKT。6次LPS处理的大鼠显示总JNK和pJNK增加。6次LPS处理的大鼠对中枢注射胰岛素(12μM/5μL,脑室内注射)诱导的摄食减少效应无反应。长期暴露于LPS(24小时)损害了胰岛素诱导的AKT磷酸化以及培养的下丘脑GT1-7细胞中转录因子叉头框蛋白O1(FoxO1)从细胞核向细胞质的转位。脑室内注射JNK抑制剂(20μM/5μL)恢复了6次LPS处理大鼠中胰岛素使IRS1和AKT磷酸化的能力。目前的数据表明,下丘脑JNK活性增加是长期暴露于内毒素期间胰岛素抵抗发展的基础。我们的研究表明,下丘脑胰岛素抵抗的发展并非必须体重增加,并且在肥胖中所见的长期低度炎症期间,阻断促炎途径可能有助于恢复胰岛素信号传导。