Selli Daniele, Fazio Gianluca, Seifert Gotthard, Di Valentin Cristiana
Dipartimento di Scienza dei Materiali, Università di Milano-Bicocca , Milano, Italy.
Technische Universität Dresden , Institut für Theoretische Chemie, D-01062 Dresden, Germany.
J Chem Theory Comput. 2017 Aug 8;13(8):3862-3873. doi: 10.1021/acs.jctc.7b00479. Epub 2017 Jul 20.
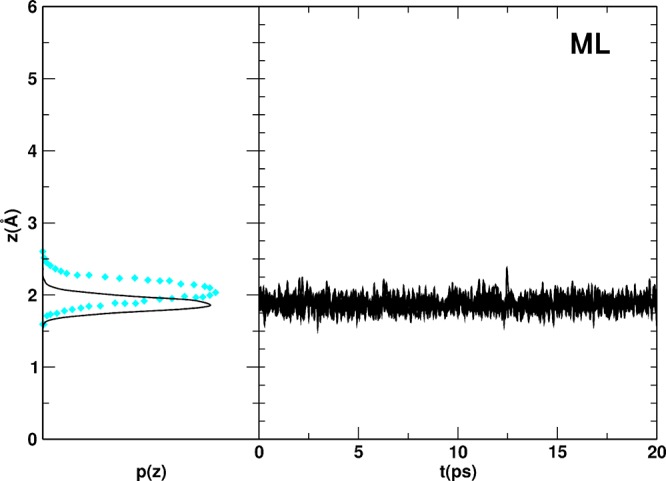
A water/(101) anatase TiO interface has been investigated with the DFT-based self-consistent-charge density functional tight-binding theory (SCC-DFTB). By comparison of the computed structural, energetic, and dynamical properties with standard DFT-GGA and experimental data, we assess the accuracy of SCC-DFTB for this prototypical solid-liquid interface. We tested different available SCC-DFTB parameters for Ti-containing compounds and, accordingly, combined them to improve the reliability of the method. To better describe water energetics, we have also introduced a modified hydrogen-bond-damping function (HBD). With this correction, equilibrium structures and adsorption energies of water on (101) anatase both for low (0.25 ML) and full (1 ML) coverages are in excellent agreement with those obtained with a higher level of theory (DFT-GGA). Furthermore, Born-Oppenheimer molecular dynamics (MD) simulations for mono-, bi-, and trilayers of water on the surface, as computed with SCC-DFTB, evidence similar ordering and energetics as DFT-GGA Car-Parrinello MD results. Finally, we have evaluated the energy barrier for the dissociation of a water molecule on the anatase (101) surface. Overall, the combined set of parameters with the HBD correction (SCC-DFTB+HBD) is shown to provide a description of the water/water/titania interface, which is very close to that obtained by standard DFT-GGA, with a remarkably reduced computational cost. Hence, this study opens the way to the future investigations on much more extended and realistic TiO/liquid water systems, which are extremely relevant for many modern technological applications.
采用基于密度泛函理论的自洽电荷密度泛函紧束缚理论(SCC-DFTB)研究了水/(101)锐钛矿TiO界面。通过将计算得到的结构、能量和动力学性质与标准密度泛函理论广义梯度近似(DFT-GGA)及实验数据进行比较,我们评估了SCC-DFTB对这种典型固液界面的准确性。我们测试了含钛化合物的不同可用SCC-DFTB参数,并相应地进行组合以提高该方法的可靠性。为了更好地描述水的能量学,我们还引入了一种改进的氢键阻尼函数(HBD)。通过这种修正,低覆盖度(0.25 ML)和全覆盖度(1 ML)下,水在(101)锐钛矿上的平衡结构和吸附能与用更高理论水平(DFT-GGA)得到的结果高度吻合。此外,用SCC-DFTB计算得到的表面单分子层、双分子层和三分子层水的玻恩-奥本海默分子动力学(MD)模拟结果表明,其有序性和能量学与DFT-GGA Car-Parrinello MD模拟结果相似。最后,我们评估了锐钛矿(101)表面水分子解离的能垒。总体而言,结合HBD修正的参数集(SCC-DFTB+HBD)能够很好地描述水/水/二氧化钛界面,与标准DFT-GGA得到的结果非常接近,且计算成本显著降低。因此,本研究为未来对更广泛、更真实的TiO/液态水体系的研究开辟了道路,这些体系与许多现代技术应用密切相关。