Pham Tuan Anh, Govoni Marco, Seidel Robert, Bradforth Stephen E, Schwegler Eric, Galli Giulia
Quantum Simulations Group, Lawrence Livermore National Laboratory, Livermore, CA 94550, USA.
Institute for Molecular Engineering, University of Chicago, Chicago, IL 60637, USA.
Sci Adv. 2017 Jun 23;3(6):e1603210. doi: 10.1126/sciadv.1603210. eCollection 2017 Jun.
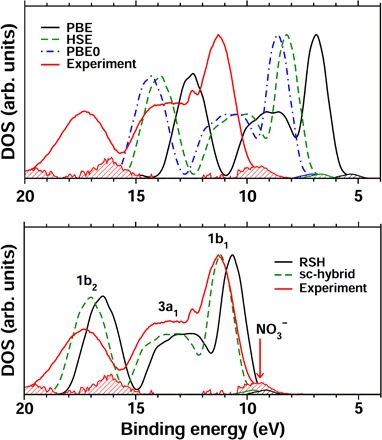
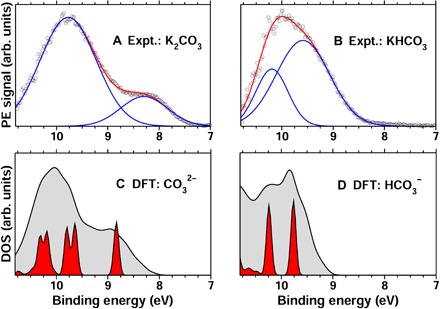
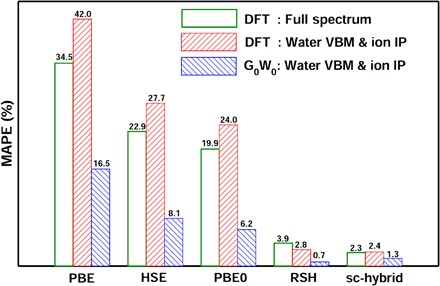
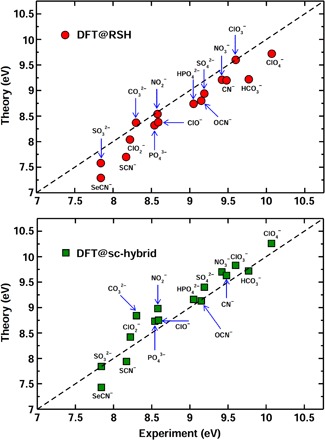
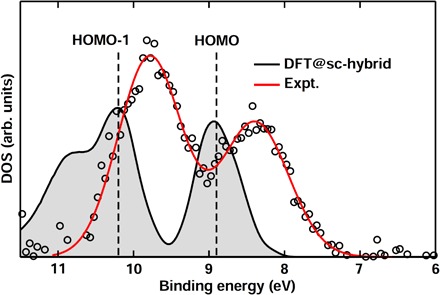
Predicting the electronic properties of aqueous liquids has been a long-standing challenge for quantum mechanical methods. However, it is a crucial step in understanding and predicting the key role played by aqueous solutions and electrolytes in a wide variety of emerging energy and environmental technologies, including battery and photoelectrochemical cell design. We propose an efficient and accurate approach to predict the electronic properties of aqueous solutions, on the basis of the combination of first-principles methods and experimental validation using state-of-the-art spectroscopic measurements. We present results of the photoelectron spectra of a broad range of solvated ions, showing that first-principles molecular dynamics simulations and electronic structure calculations using dielectric hybrid functionals provide a quantitative description of the electronic properties of the solvent and solutes, including excitation energies. The proposed computational framework is general and applicable to other liquids, thereby offering great promise in understanding and engineering solutions and liquid electrolytes for a variety of important energy technologies.
对量子力学方法而言,预测水性液体的电子性质一直是一项长期挑战。然而,这是理解和预测水溶液及电解质在包括电池和光电化学电池设计在内的各种新兴能源与环境技术中所起关键作用的关键一步。我们提出了一种高效且准确的方法来预测水溶液的电子性质,该方法基于第一性原理方法与使用最先进光谱测量进行的实验验证相结合。我们展示了一系列溶剂化离子的光电子能谱结果,表明使用介电混合泛函的第一性原理分子动力学模拟和电子结构计算能够对溶剂和溶质的电子性质进行定量描述,包括激发能。所提出的计算框架具有通用性,适用于其他液体,从而在理解和设计用于各种重要能源技术的溶液及液体电解质方面展现出巨大潜力。