Shukla Surendra K, Purohit Vinee, Mehla Kamiya, Gunda Venugopal, Chaika Nina V, Vernucci Enza, King Ryan J, Abrego Jaime, Goode Gennifer D, Dasgupta Aneesha, Illies Alysha L, Gebregiworgis Teklab, Dai Bingbing, Augustine Jithesh J, Murthy Divya, Attri Kuldeep S, Mashadova Oksana, Grandgenett Paul M, Powers Robert, Ly Quan P, Lazenby Audrey J, Grem Jean L, Yu Fang, Matés José M, Asara John M, Kim Jung-Whan, Hankins Jordan H, Weekes Colin, Hollingsworth Michael A, Serkova Natalie J, Sasson Aaron R, Fleming Jason B, Oliveto Jennifer M, Lyssiotis Costas A, Cantley Lewis C, Berim Lyudmyla, Singh Pankaj K
Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE 68198-5950, USA.
Eppley Institute for Research in Cancer and Allied Diseases, University of Nebraska Medical Center, Omaha, NE 68198-5950, USA; Department of Pathology and Microbiology, University of Nebraska Medical Center, Omaha, NE 68198, USA.
Cancer Cell. 2017 Jul 10;32(1):71-87.e7. doi: 10.1016/j.ccell.2017.06.004.
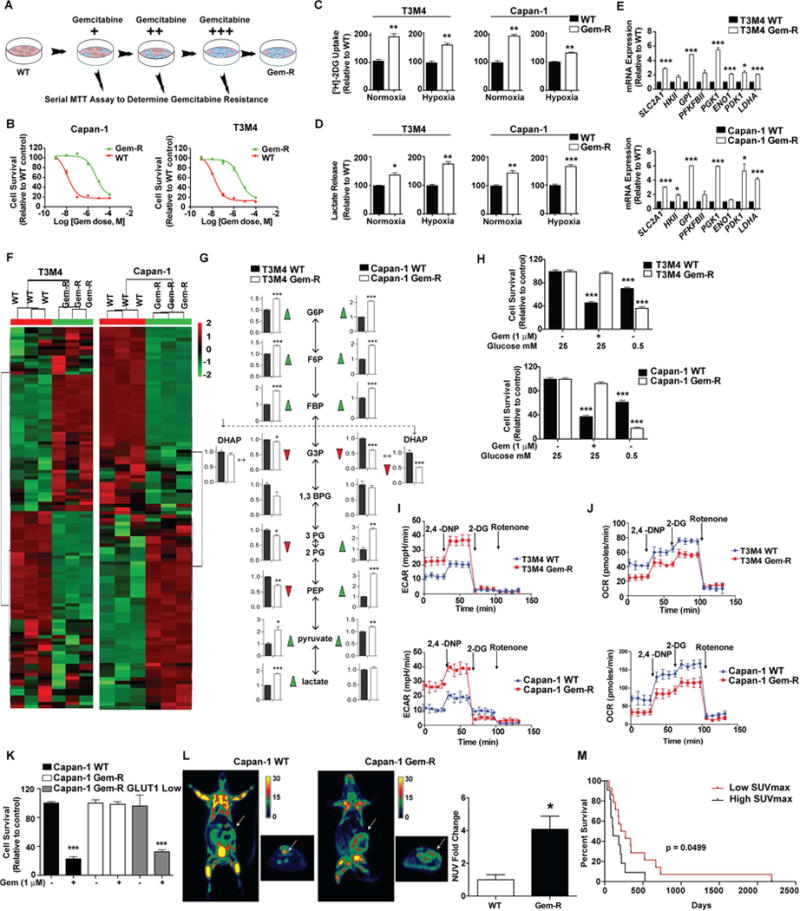
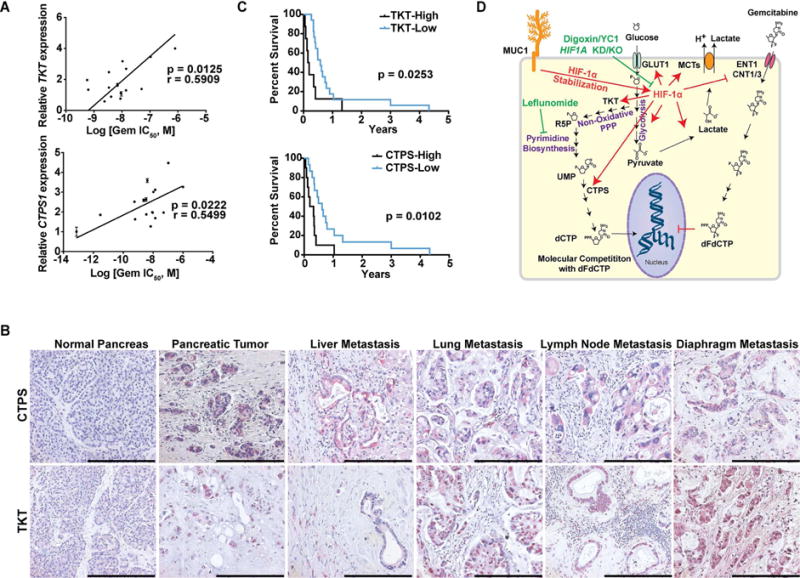
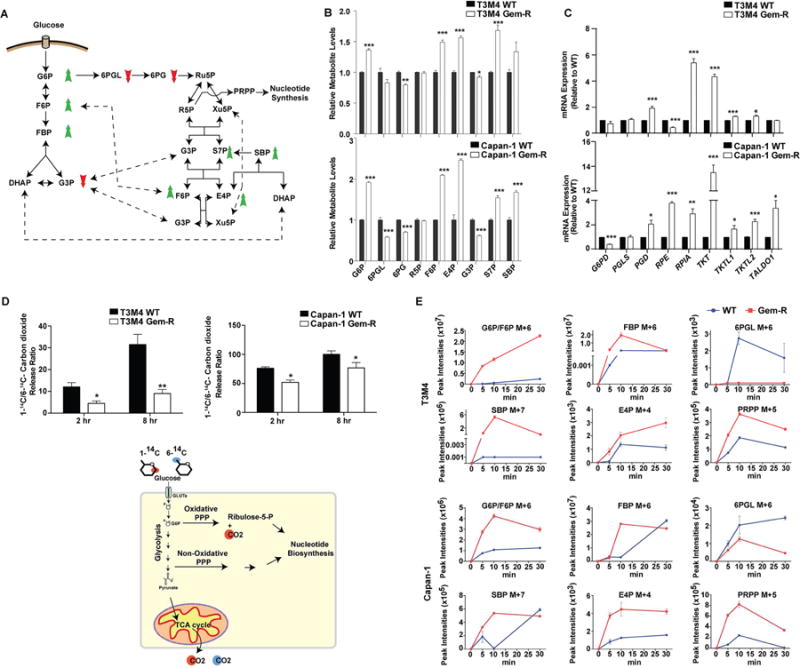
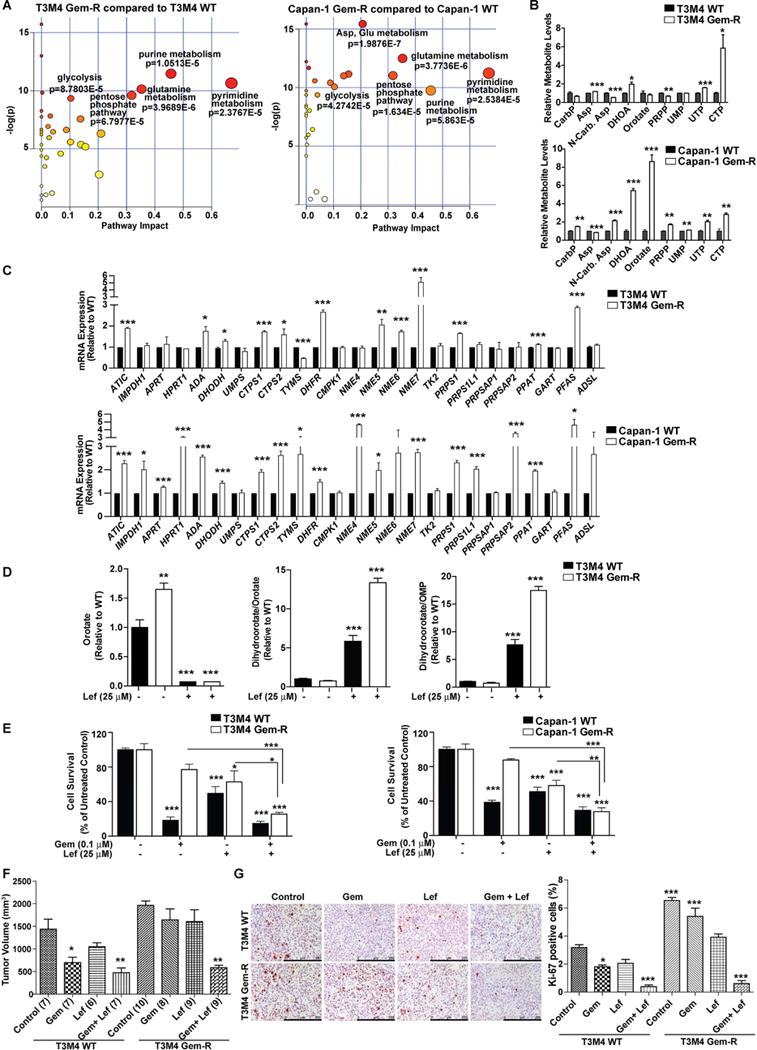
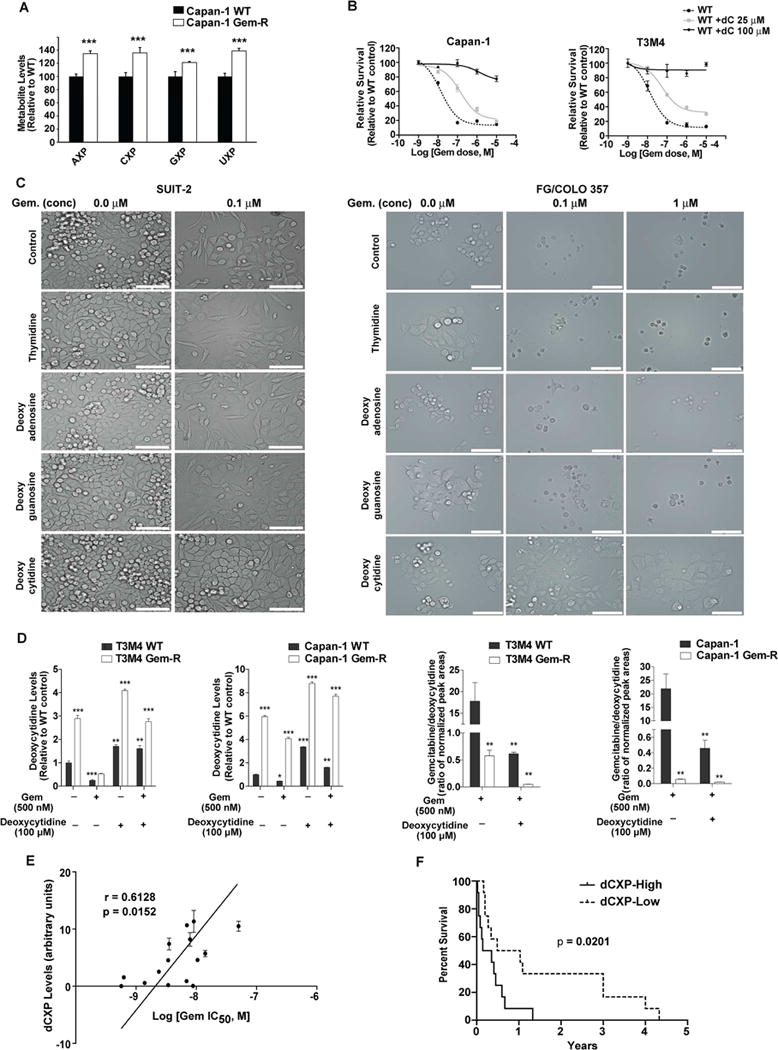
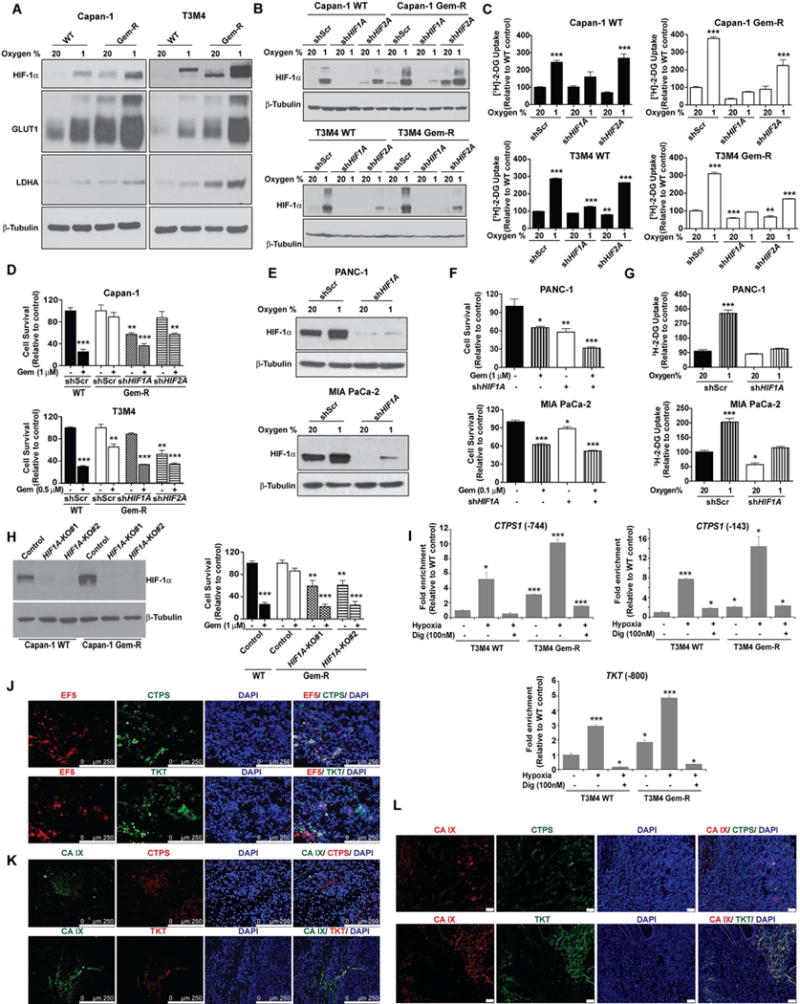
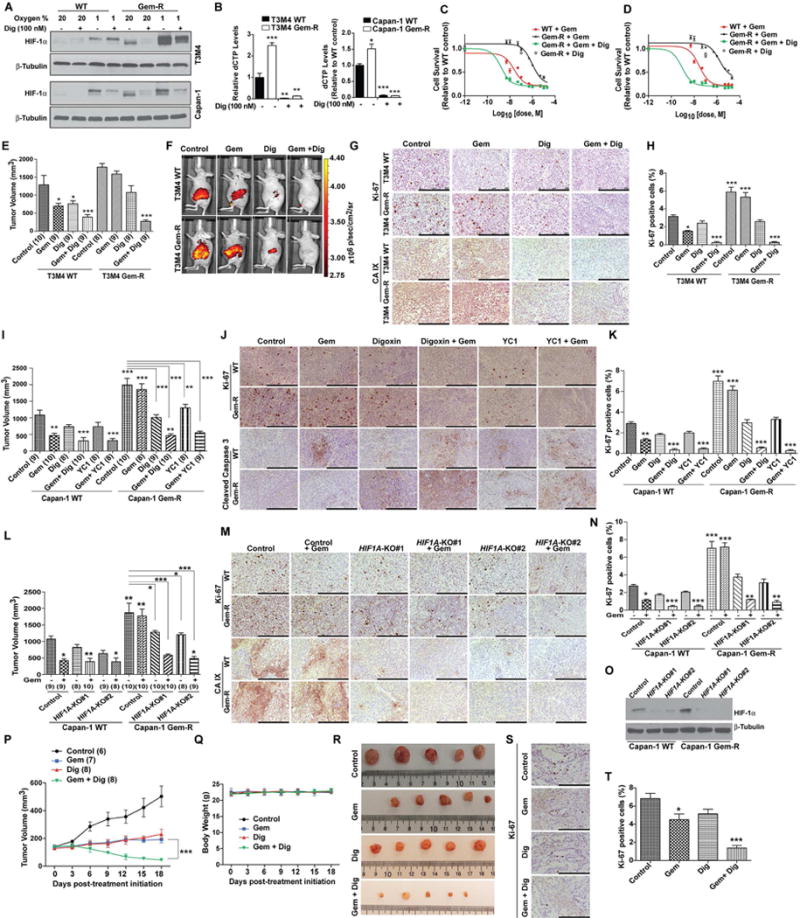
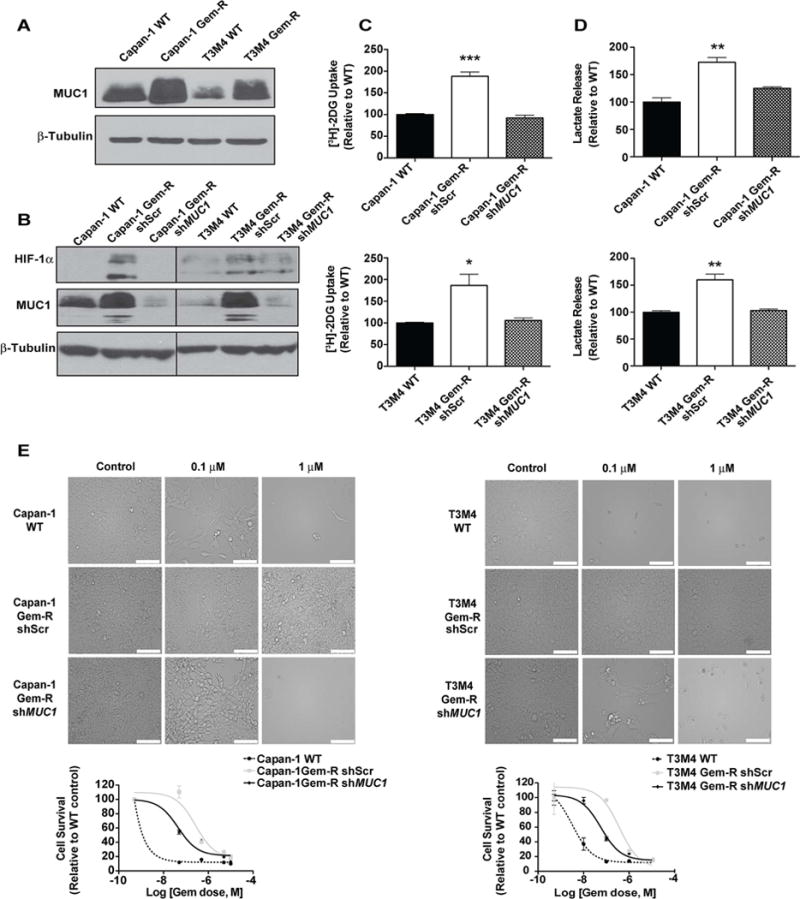
Poor response to cancer therapy due to resistance remains a clinical challenge. The present study establishes a widely prevalent mechanism of resistance to gemcitabine in pancreatic cancer, whereby increased glycolytic flux leads to glucose addiction in cancer cells and a corresponding increase in pyrimidine biosynthesis to enhance the intrinsic levels of deoxycytidine triphosphate (dCTP). Increased levels of dCTP diminish the effective levels of gemcitabine through molecular competition. We also demonstrate that MUC1-regulated stabilization of hypoxia inducible factor-1α (HIF-1α) mediates such metabolic reprogramming. Targeting HIF-1α or de novo pyrimidine biosynthesis, in combination with gemcitabine, strongly diminishes tumor burden. Finally, reduced expression of TKT and CTPS, which regulate flux into pyrimidine biosynthesis, correlates with better prognosis in pancreatic cancer patients on fluoropyrimidine analogs.
由于耐药性导致的癌症治疗反应不佳仍然是一项临床挑战。本研究确立了胰腺癌中一种广泛存在的吉西他滨耐药机制,即糖酵解通量增加导致癌细胞对葡萄糖成瘾,同时嘧啶生物合成相应增加,以提高三磷酸脱氧胞苷(dCTP)的内在水平。dCTP水平升高通过分子竞争降低了吉西他滨的有效水平。我们还证明,MUC1调节的缺氧诱导因子-1α(HIF-1α)的稳定介导了这种代谢重编程。靶向HIF-1α或从头嘧啶生物合成,并与吉西他滨联合使用,可显著减轻肿瘤负担。最后,调节嘧啶生物合成通量的TKT和CTPS表达降低,与接受氟嘧啶类似物治疗的胰腺癌患者的较好预后相关。