Parzefall Thomas, Frohne Alexandra, Koenighofer Martin, Kirchnawy Andreas, Streubel Berthold, Schoefer Christian, Gstoettner Wolfgang, Frei Klemens, Lucas Trevor
Department of Otorhinolaryngology, Head and Neck Surgery, Medical University of Vienna, Waehringer Guertel 18-20, 1090, Vienna, Austria.
Department for Cell and Developmental Biology, Orphan disease genetics group, Center for Anatomy and Cell Biology, Medical University of Vienna, Vienna, Austria.
Wien Klin Wochenschr. 2018 May;130(9-10):299-306. doi: 10.1007/s00508-017-1230-y. Epub 2017 Jul 21.
Non-syndromic autosomal dominant hearing impairment is characteristically postlingual in onset. Genetic diagnostics are essential for genetic counselling, disease prognosis and understanding of the molecular mechanisms of disease. To date, 36 causative genes have been identified, many in only individual families. Gene selection for genetic screening by traditional methods and genetic diagnosis in autosomal dominant patients has therefore been fraught with difficulty. Whole-exome sequencing provides a powerful tool to analyze all protein-coding genomic regions in parallel, thus allowing the comprehensive screening of all known genes and associated alterations.
In this study, a previously undiagnosed late-onset progressive autosomal dominant hearing loss in an Austrian family was investigated by means of whole-exome sequencing. Results were confirmed by Sanger sequencing.
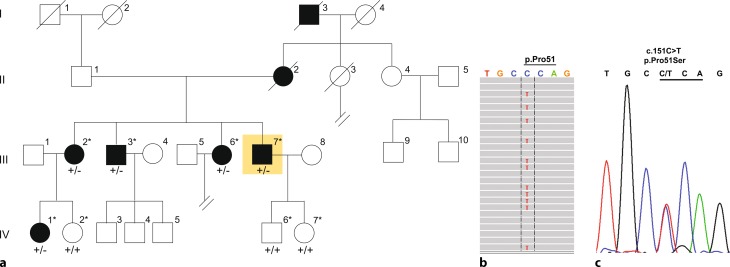
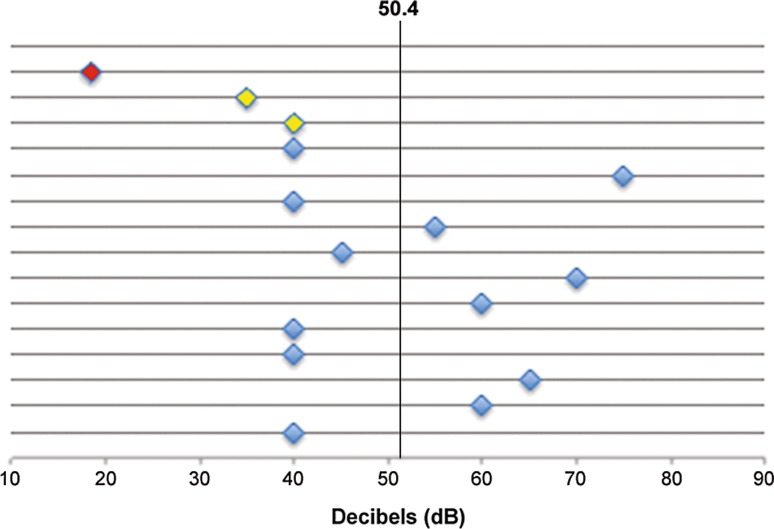
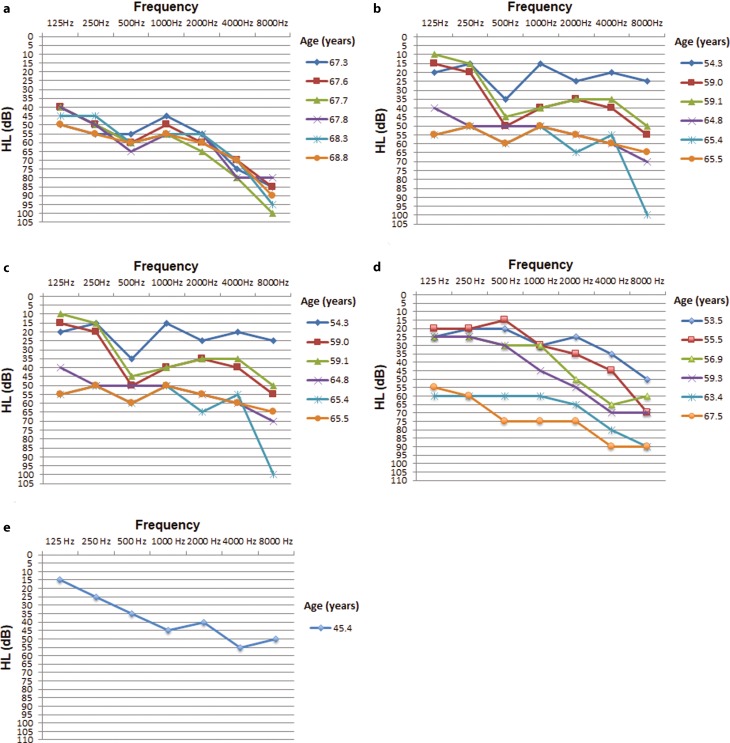
A previously described c.151C>T missense (p.Pro51Ser) mutation in the LCCL (limulus factor C, cochlin, late gestation lung protein Lgl1) domain of the cochlin gene (COCH) was identified as causative and segregated with disease in five members of the family. Molecular diagnostics led to the decision to perform cochlear implantation in an index patient who subsequently showed excellent postoperative auditory performance. The c.151C>T mutation was not found in 18 screened Austrian families with autosomal dominant hearing loss but was represented alongside other known pathogenic mutant COCH alleles in the Genome Aggregation Database (gnomAD) in European populations. A combined allele frequency of 0.000128 implies an orphan disease frequency for COCH-induced hearing loss of 1:3900 in Europe.
Exome sequencing successfully resolved the genetic diagnosis in a family suffering from autosomal dominant hearing impairment and allowed prediction of purported auditory outcome after cochlear implantation in an index patient. Personalized treatment approaches based on the molecular mechanisms of disease may become increasingly important in the future.
非综合征性常染色体显性遗传性听力障碍的典型发病时间为语言发育期之后。基因诊断对于遗传咨询、疾病预后以及了解疾病的分子机制至关重要。迄今为止,已鉴定出36个致病基因,其中许多仅在个别家族中被发现。因此,通过传统方法进行遗传筛查以及对常染色体显性患者进行基因诊断时,基因选择一直存在困难。全外显子测序提供了一个强大的工具,可以同时分析所有蛋白质编码基因组区域,从而能够对所有已知基因和相关变异进行全面筛查。
在本研究中,通过全外显子测序对一个奥地利家族中先前未确诊的迟发性进行性常染色体显性听力损失进行了调查。结果通过桑格测序得到证实。
在耳蜗蛋白基因(COCH)的LCCL(鲎因子C、耳蜗蛋白、妊娠晚期肺蛋白Lgl1)结构域中,先前描述的c.151C>T错义突变(p.Pro51Ser)被确定为致病突变,并在该家族的五名成员中与疾病共分离。分子诊断促使决定对一名索引患者进行人工耳蜗植入,该患者术后听觉表现极佳。在18个经筛查的患有常染色体显性听力损失的奥地利家族中未发现c.151C>T突变,但在欧洲人群的基因组聚合数据库(gnomAD)中,该突变与其他已知的致病性COCH突变等位基因同时存在。合并等位基因频率为0.000128,这意味着在欧洲,由COCH引起的听力损失的孤儿病频率为1:3900。
外显子测序成功解决了一个患有常染色体显性听力障碍家族的基因诊断问题,并能够预测索引患者人工耳蜗植入后的听觉结果。基于疾病分子机制的个性化治疗方法在未来可能会变得越来越重要。