Yan Koon-Kiu, Lou Shaoke, Gerstein Mark
Program in Computational Biology and Bioinformatics, Yale University, New Haven, CT, United States of America.
Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT, United States of America.
PLoS Comput Biol. 2017 Jul 24;13(7):e1005647. doi: 10.1371/journal.pcbi.1005647. eCollection 2017 Jul.
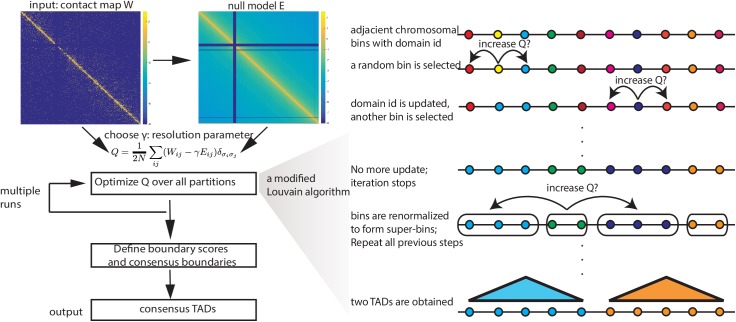
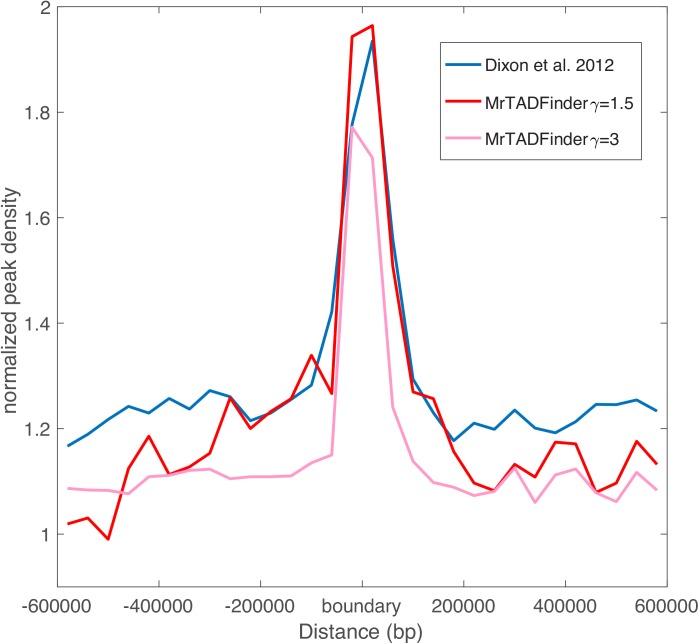
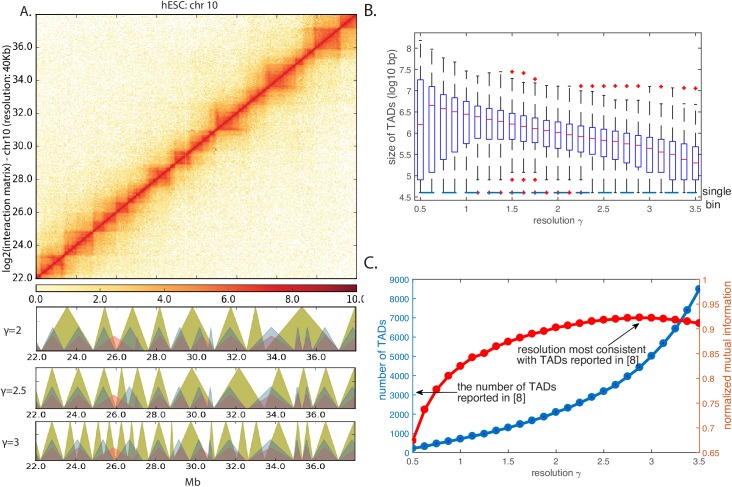
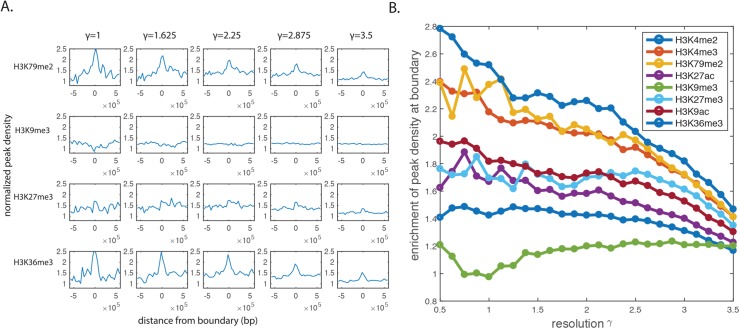
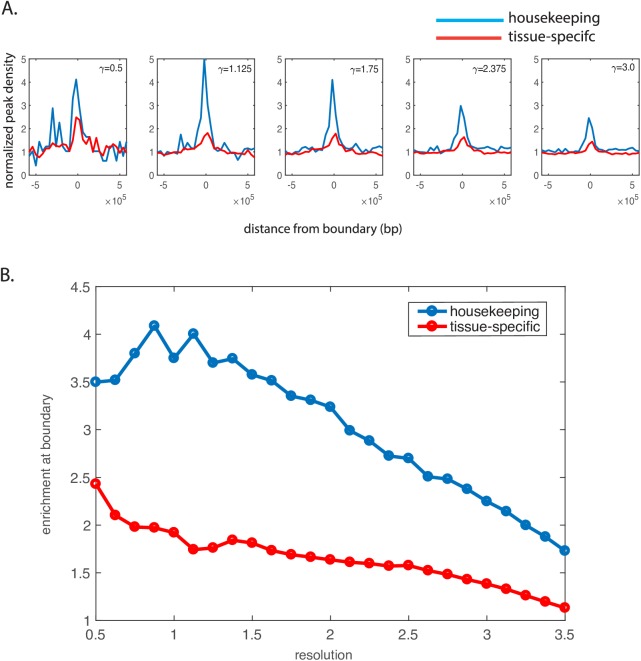
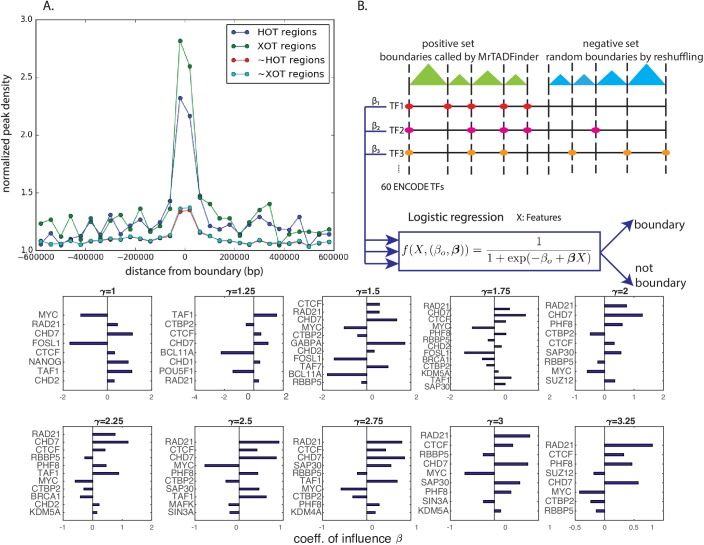
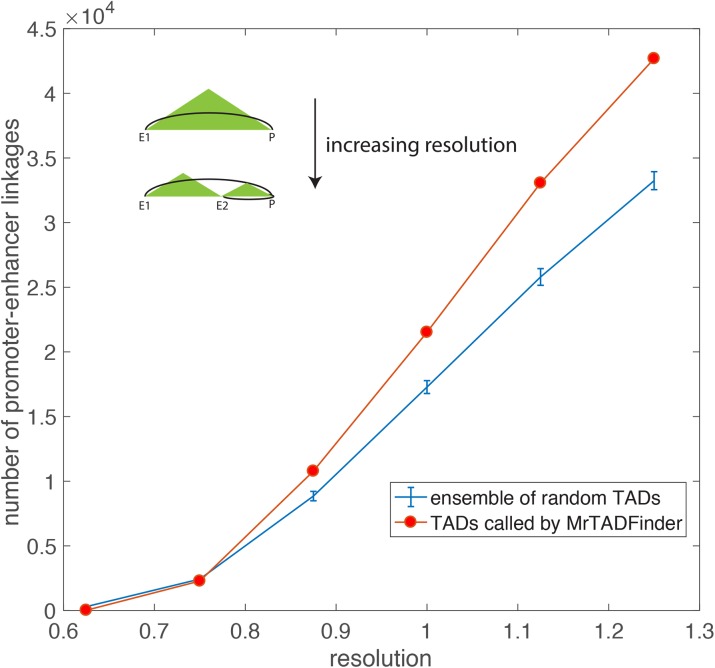
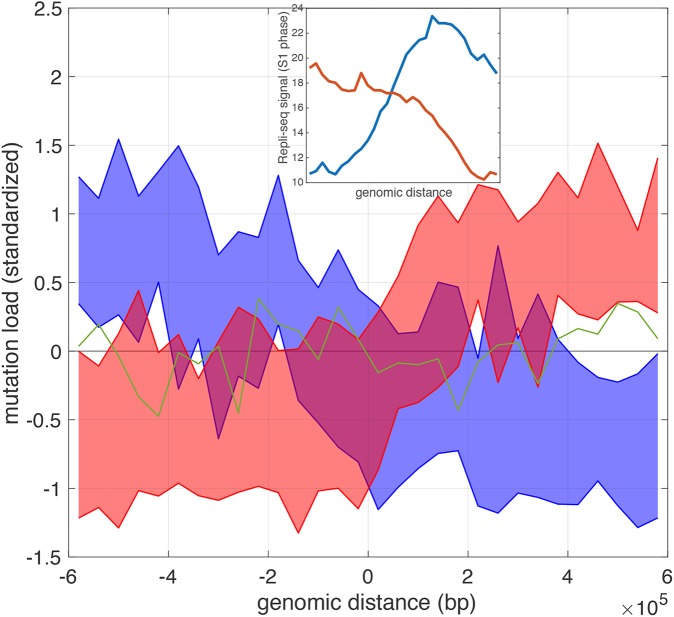
Genome-wide proximity ligation based assays such as Hi-C have revealed that eukaryotic genomes are organized into structural units called topologically associating domains (TADs). From a visual examination of the chromosomal contact map, however, it is clear that the organization of the domains is not simple or obvious. Instead, TADs exhibit various length scales and, in many cases, a nested arrangement. Here, by exploiting the resemblance between TADs in a chromosomal contact map and densely connected modules in a network, we formulate TAD identification as a network optimization problem and propose an algorithm, MrTADFinder, to identify TADs from intra-chromosomal contact maps. MrTADFinder is based on the network-science concept of modularity. A key component of it is deriving an appropriate background model for contacts in a random chain, by numerically solving a set of matrix equations. The background model preserves the observed coverage of each genomic bin as well as the distance dependence of the contact frequency for any pair of bins exhibited by the empirical map. Also, by introducing a tunable resolution parameter, MrTADFinder provides a self-consistent approach for identifying TADs at different length scales, hence the acronym "Mr" standing for Multiple Resolutions. We then apply MrTADFinder to various Hi-C datasets. The identified domain boundaries are marked by characteristic signatures in chromatin marks and transcription factors (TF) that are consistent with earlier work. Moreover, by calling TADs at different length scales, we observe that boundary signatures change with resolution, with different chromatin features having different characteristic length scales. Furthermore, we report an enrichment of HOT (high-occupancy target) regions near TAD boundaries and investigate the role of different TFs in determining boundaries at various resolutions. To further explore the interplay between TADs and epigenetic marks, as tumor mutational burden is known to be coupled to chromatin structure, we examine how somatic mutations are distributed across boundaries and find a clear stepwise pattern. Overall, MrTADFinder provides a novel computational framework to explore the multi-scale structures in Hi-C contact maps.
基于全基因组邻近连接的分析方法,如Hi-C,已经揭示真核生物基因组被组织成称为拓扑相关结构域(TADs)的结构单元。然而,从染色体接触图谱的视觉检查来看,很明显这些结构域的组织并不简单或明显。相反,TADs表现出各种长度尺度,并且在许多情况下是嵌套排列的。在这里,通过利用染色体接触图谱中的TADs与网络中紧密连接的模块之间的相似性,我们将TAD识别表述为一个网络优化问题,并提出一种算法MrTADFinder,用于从染色体内接触图谱中识别TADs。MrTADFinder基于模块化的网络科学概念。它的一个关键组成部分是通过数值求解一组矩阵方程,为随机链中的接触推导一个合适的背景模型。该背景模型保留了每个基因组区间的观察覆盖范围以及经验图谱中任意一对区间接触频率的距离依赖性。此外,通过引入一个可调分辨率参数,MrTADFinder提供了一种自洽的方法来识别不同长度尺度的TADs,因此首字母缩写“Mr”代表多分辨率。然后我们将MrTADFinder应用于各种Hi-C数据集。识别出的结构域边界由染色质标记和转录因子(TF)中的特征信号标记,这与早期工作一致。此外,通过在不同长度尺度上调用TADs,我们观察到边界信号随分辨率变化,不同的染色质特征具有不同的特征长度尺度。此外,我们报告了TAD边界附近高占用靶点(HOT)区域的富集,并研究了不同TF在确定不同分辨率下边界的作用。为了进一步探索TADs与表观遗传标记之间的相互作用,由于已知肿瘤突变负担与染色质结构相关,我们研究了体细胞突变如何分布在边界上,并发现了一种明显的阶梯模式。总体而言,MrTADFinder提供了一个新颖的计算框架来探索Hi-C接触图谱中的多尺度结构。