Omidi Saeed, Zavolan Mihaela, Pachkov Mikhail, Breda Jeremie, Berger Severin, van Nimwegen Erik
Biozentrum, University of Basel, Basel, Switzerland.
Swiss Institute of Bioinformatics, Basel, Switzerland.
PLoS Comput Biol. 2017 Jul 28;13(7):e1005176. doi: 10.1371/journal.pcbi.1005176. eCollection 2017 Jul.
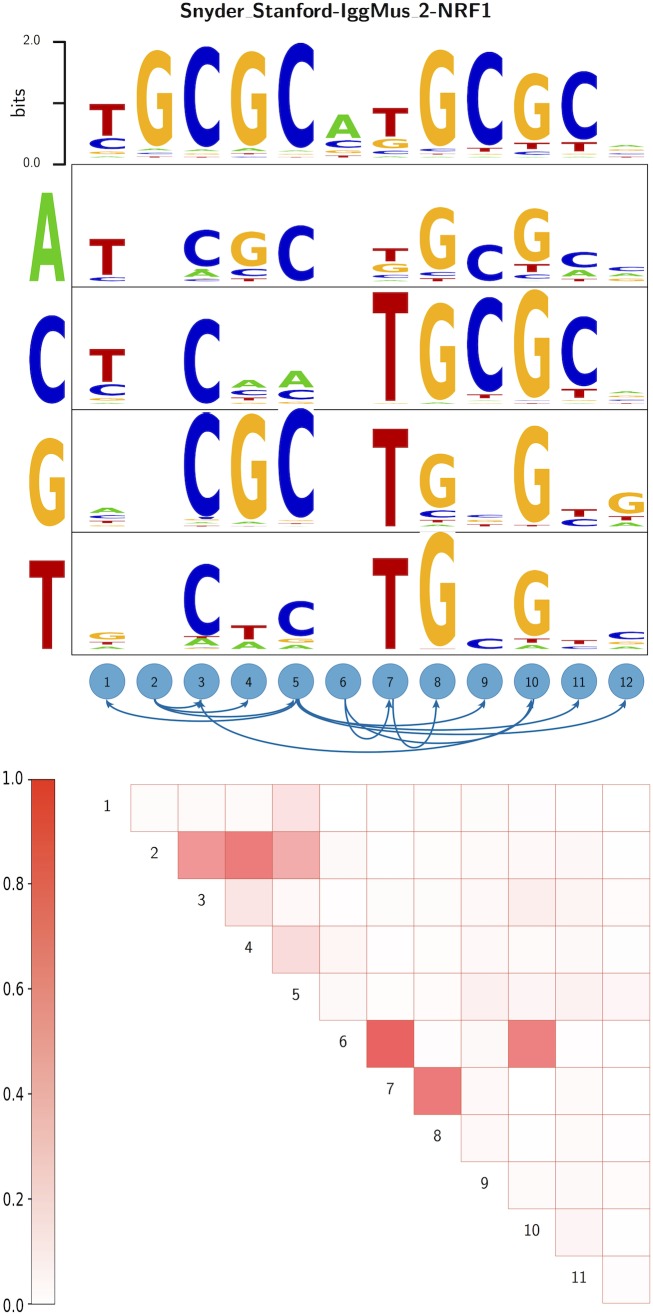
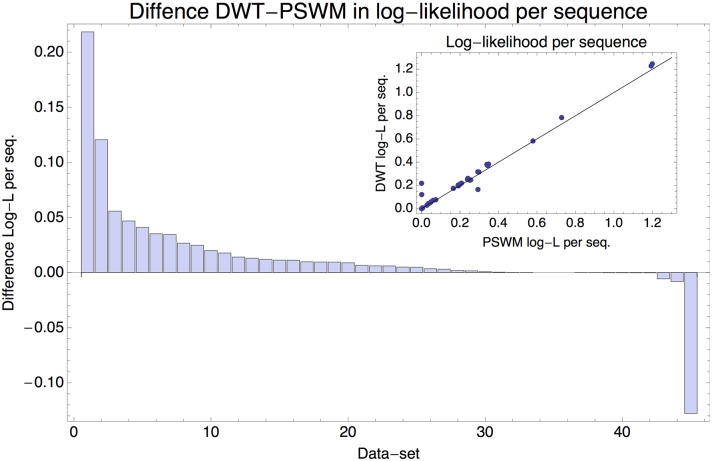
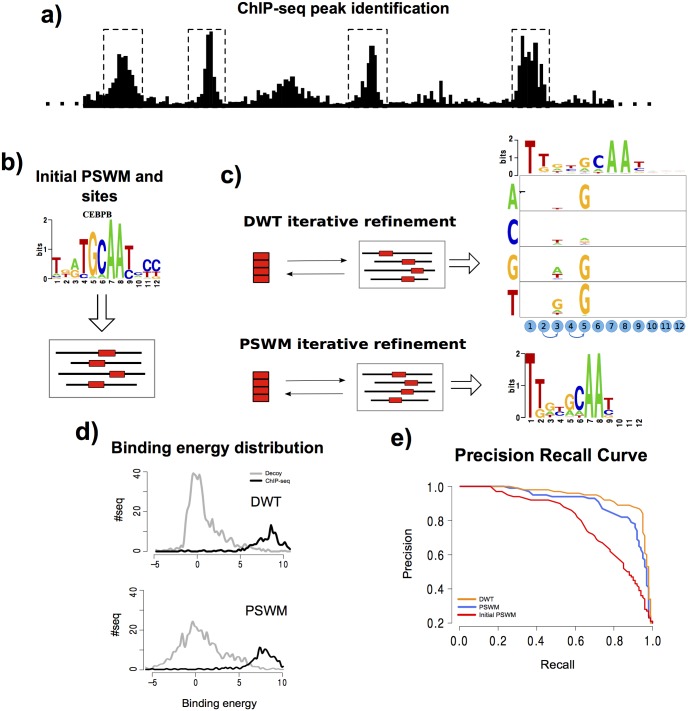
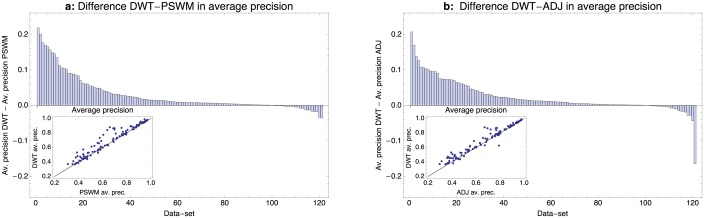
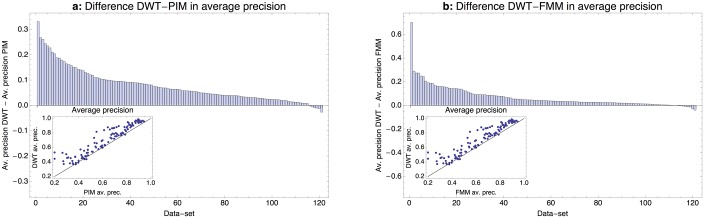
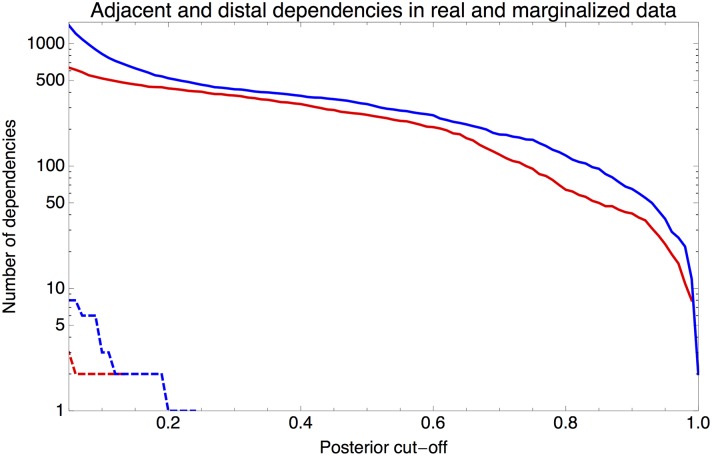
Gene regulatory networks are ultimately encoded by the sequence-specific binding of (TFs) to short DNA segments. Although it is customary to represent the binding specificity of a TF by a position-specific weight matrix (PSWM), which assumes each position within a site contributes independently to the overall binding affinity, evidence has been accumulating that there can be significant dependencies between positions. Unfortunately, methodological challenges have so far hindered the development of a practical and generally-accepted extension of the PSWM model. On the one hand, simple models that only consider dependencies between nearest-neighbor positions are easy to use in practice, but fail to account for the distal dependencies that are observed in the data. On the other hand, models that allow for arbitrary dependencies are prone to overfitting, requiring regularization schemes that are difficult to use in practice for non-experts. Here we present a new regulatory motif model, called dinucleotide weight tensor (DWT), that incorporates arbitrary pairwise dependencies between positions in binding sites, rigorously from first principles, and free from tunable parameters. We demonstrate the power of the method on a large set of ChIP-seq data-sets, showing that DWTs outperform both PSWMs and motif models that only incorporate nearest-neighbor dependencies. We also demonstrate that DWTs outperform two previously proposed methods. Finally, we show that DWTs inferred from ChIP-seq data also outperform PSWMs on HT-SELEX data for the same TF, suggesting that DWTs capture inherent biophysical properties of the interactions between the DNA binding domains of TFs and their binding sites. We make a suite of DWT tools available at dwt.unibas.ch, that allow users to automatically perform 'motif finding', i.e. the inference of DWT motifs from a set of sequences, binding site prediction with DWTs, and visualization of DWT 'dilogo' motifs.
基因调控网络最终由转录因子(TFs)与短DNA片段的序列特异性结合所编码。虽然通常用位置特异性权重矩阵(PSWM)来表示转录因子的结合特异性,该矩阵假设位点内的每个位置对整体结合亲和力有独立贡献,但越来越多的证据表明,位置之间可能存在显著的依赖性。不幸的是,方法上的挑战迄今为止阻碍了PSWM模型实用且被广泛接受的扩展的发展。一方面,仅考虑最近邻位置之间依赖性的简单模型在实践中易于使用,但无法解释数据中观察到的远端依赖性。另一方面,允许任意依赖性的模型容易过度拟合,需要非专业人员在实践中难以使用的正则化方案。在这里,我们提出了一种新的调控基序模型,称为二核苷酸权重张量(DWT),它从第一原理严格地纳入了结合位点中位置之间的任意成对依赖性,并且没有可调参数。我们在大量的ChIP-seq数据集上展示了该方法的强大功能,表明DWT优于PSWM和仅纳入最近邻依赖性的基序模型。我们还证明DWT优于两种先前提出的方法。最后,我们表明从ChIP-seq数据推断出的DWT在相同转录因子的HT-SELEX数据上也优于PSWM,这表明DWT捕获了转录因子的DNA结合结构域与其结合位点之间相互作用的固有生物物理特性。我们在dwt.unibas.ch上提供了一套DWT工具,允许用户自动执行“基序查找”,即从一组序列中推断DWT基序、使用DWT进行结合位点预测以及可视化DWT“双核苷酸”基序。