Crompton Michael, Purnell Tom, Tyrer Hayley E, Parker Andrew, Ball Greg, Hardisty-Hughes Rachel E, Gale Richard, Williams Debbie, Dean Charlotte H, Simon Michelle M, Mallon Ann-Marie, Wells Sara, Bhutta Mahmood F, Burton Martin J, Tateossian Hilda, Brown Steve D M
Mammalian Genetics Unit, MRC Harwell Institute, Harwell, Oxfordshire, United Kingdom.
Nuffield Department of Surgical Sciences, University of Oxford, Oxford, Oxfordshire, United Kingdom.
PLoS Genet. 2017 Aug 14;13(8):e1006969. doi: 10.1371/journal.pgen.1006969. eCollection 2017 Aug.
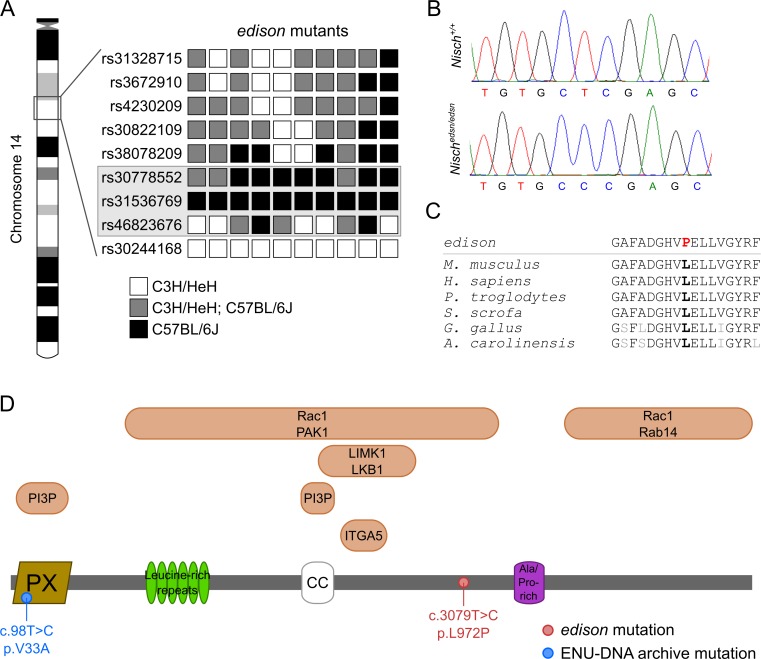
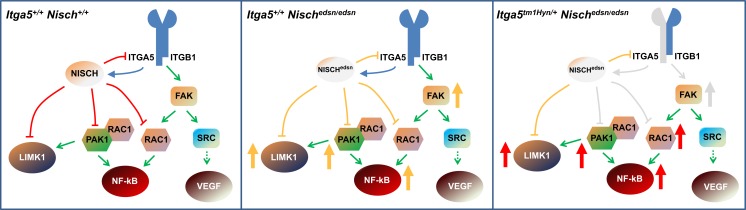
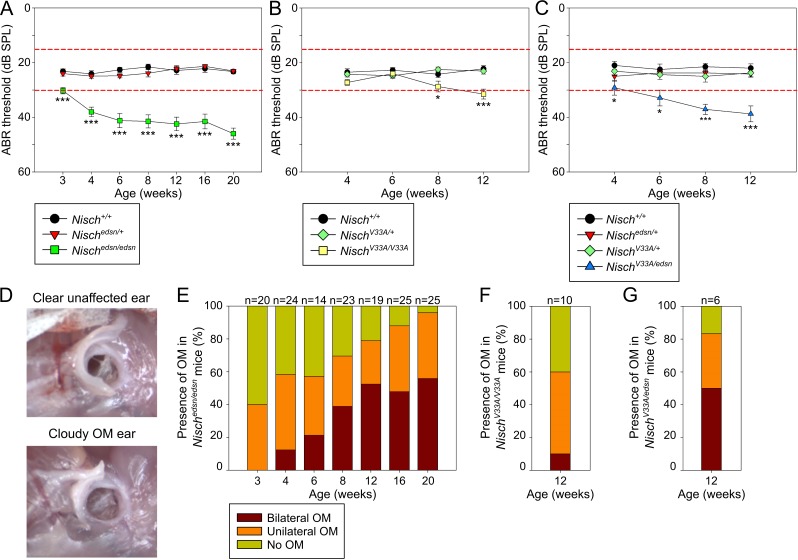
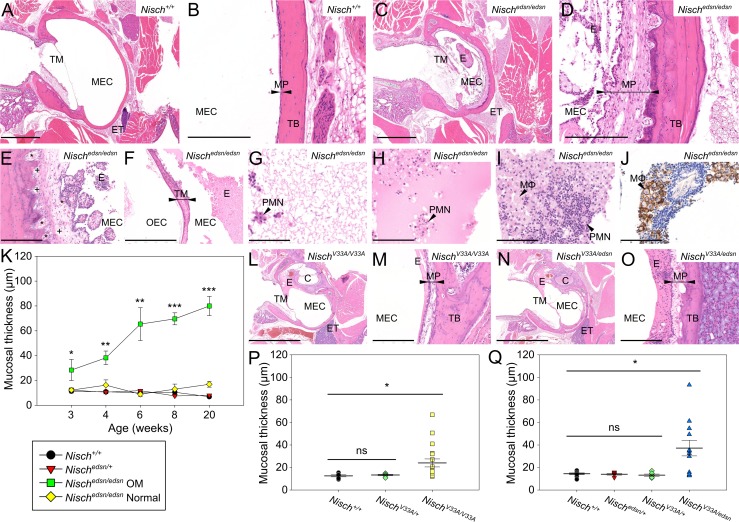
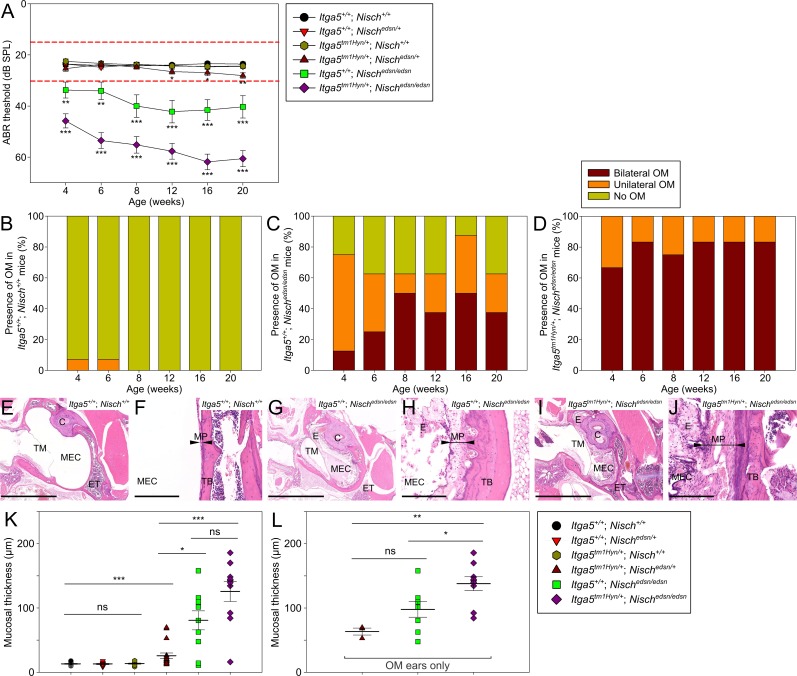
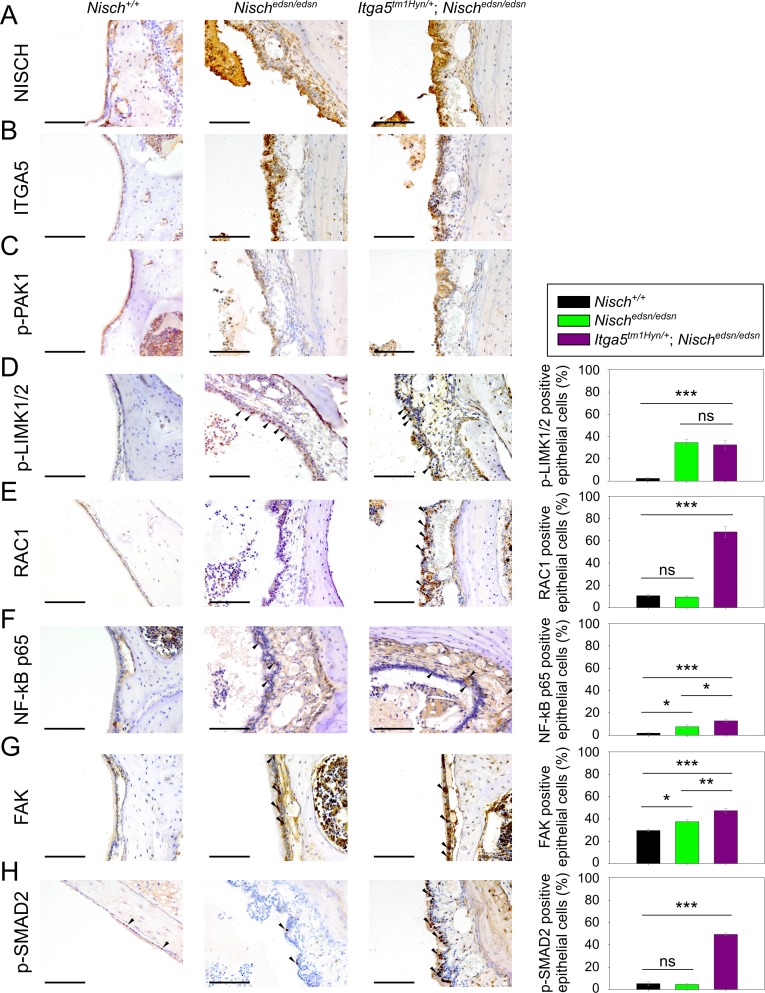
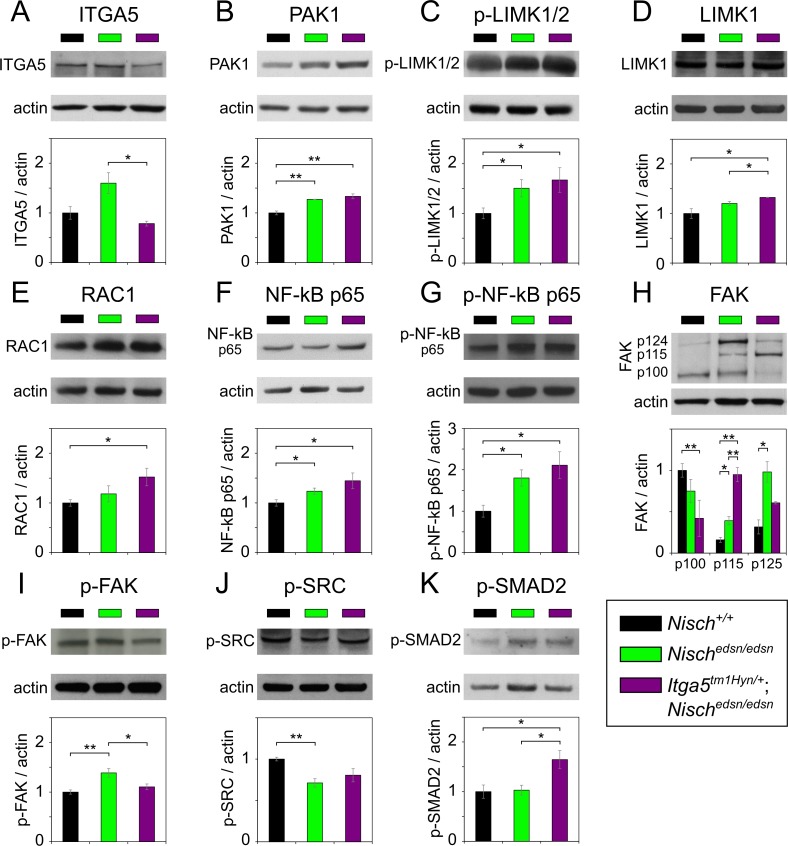
Otitis media (OM), inflammation of the middle ear (ME), is a common cause of conductive hearing impairment. Despite the importance of the disease, the aetiology of chronic and recurrent forms of middle ear inflammatory disease remains poorly understood. Studies of the human population suggest that there is a significant genetic component predisposing to the development of chronic OM, although the underlying genes are largely unknown. Using N-ethyl-N-nitrosourea mutagenesis we identified a recessive mouse mutant, edison, that spontaneously develops a conductive hearing loss due to chronic OM. The causal mutation was identified as a missense change, L972P, in the Nischarin (NISCH) gene. edison mice develop a serous or granulocytic effusion, increasingly macrophage and neutrophil rich with age, along with a thickened, inflamed mucoperiosteum. We also identified a second hypomorphic allele, V33A, with only modest increases in auditory thresholds and reduced incidence of OM. NISCH interacts with several proteins, including ITGA5 that is thought to have a role in modulating VEGF-induced angiogenesis and vascularization. We identified a significant genetic interaction between Nisch and Itga5; mice heterozygous for Itga5-null and homozygous for edison mutations display a significantly increased penetrance and severity of chronic OM. In order to understand the pathological mechanisms underlying the OM phenotype, we studied interacting partners to NISCH along with downstream signalling molecules in the middle ear epithelia of edison mouse. Our analysis implicates PAK1 and RAC1, and downstream signalling in LIMK1 and NF-κB pathways in the development of chronic OM.
中耳炎(OM),即中耳(ME)的炎症,是传导性听力障碍的常见原因。尽管该疾病很重要,但慢性和复发性中耳炎性疾病的病因仍知之甚少。对人类群体的研究表明,虽然潜在基因大多未知,但存在显著的遗传因素易导致慢性中耳炎的发生。利用N-乙基-N-亚硝基脲诱变,我们鉴定出一个隐性小鼠突变体,爱迪生(edison),它由于慢性中耳炎而自发出现传导性听力损失。因果突变被确定为Nischarin(NISCH)基因中的一个错义变化,L972P。爱迪生小鼠会出现浆液性或粒细胞性渗出,随着年龄增长,巨噬细胞和中性粒细胞越来越多,同时伴有增厚、发炎的黏骨膜。我们还鉴定出第二个低表达等位基因,V33A,其仅使听觉阈值有适度升高,且中耳炎发病率降低。NISCH与几种蛋白质相互作用,包括被认为在调节VEGF诱导的血管生成和血管化中起作用的ITGA5。我们确定了Nisch和Itga5之间存在显著的遗传相互作用;Itga5基因敲除杂合且爱迪生突变纯合的小鼠显示慢性中耳炎的外显率和严重程度显著增加。为了了解中耳炎表型背后的病理机制,我们研究了爱迪生小鼠中耳上皮中与NISCH相互作用的伙伴以及下游信号分子。我们的分析表明PAK1和RAC1以及LIMK1和NF-κB途径中的下游信号传导与慢性中耳炎的发展有关。