Department of Cardiology, First Affiliated Hospital, Nanchang University, Nanchang 330006, Jiangxi Province, China.
Department of Cardiology, Loudi Central Hospital, Loudi 417000, Hunan Province, China.
Biosci Rep. 2017 Sep 7;37(5). doi: 10.1042/BSR20171049. Print 2017 Oct 31.
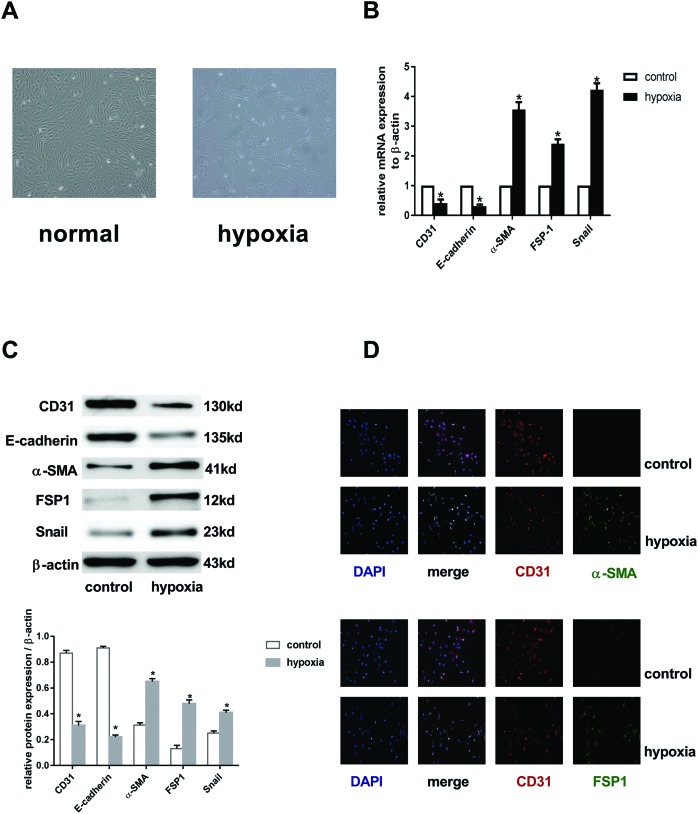
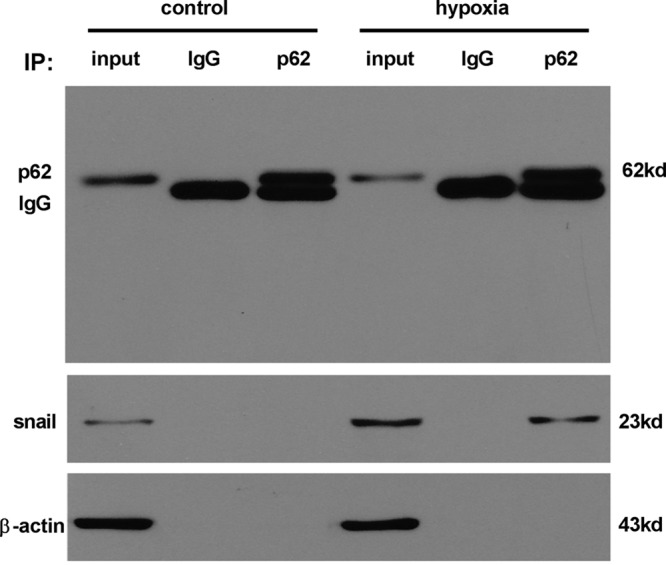
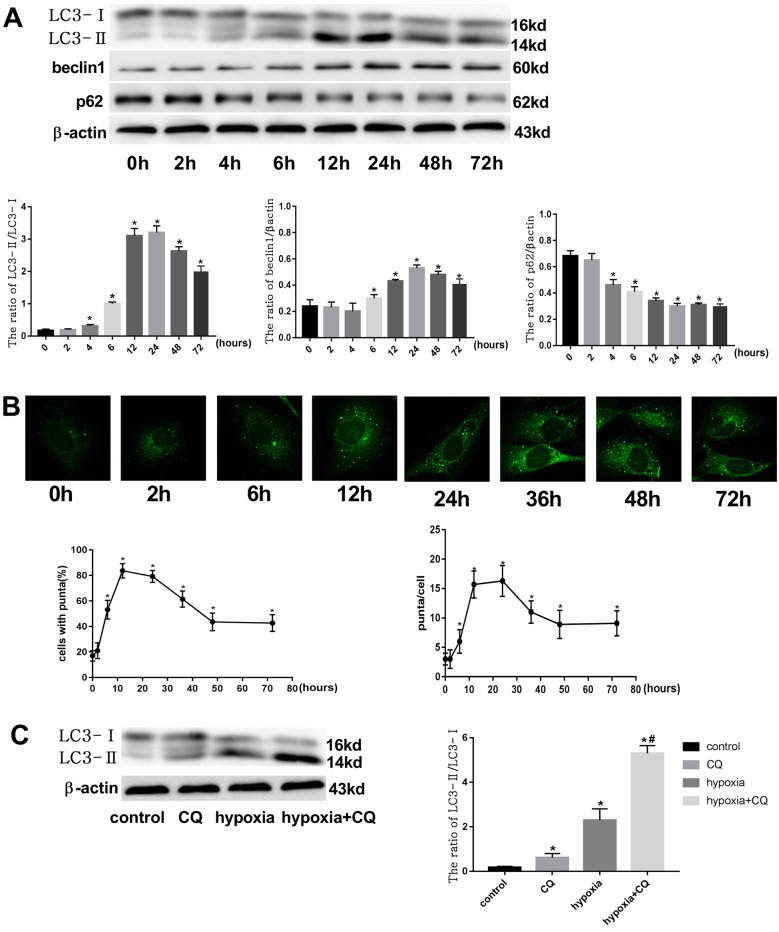
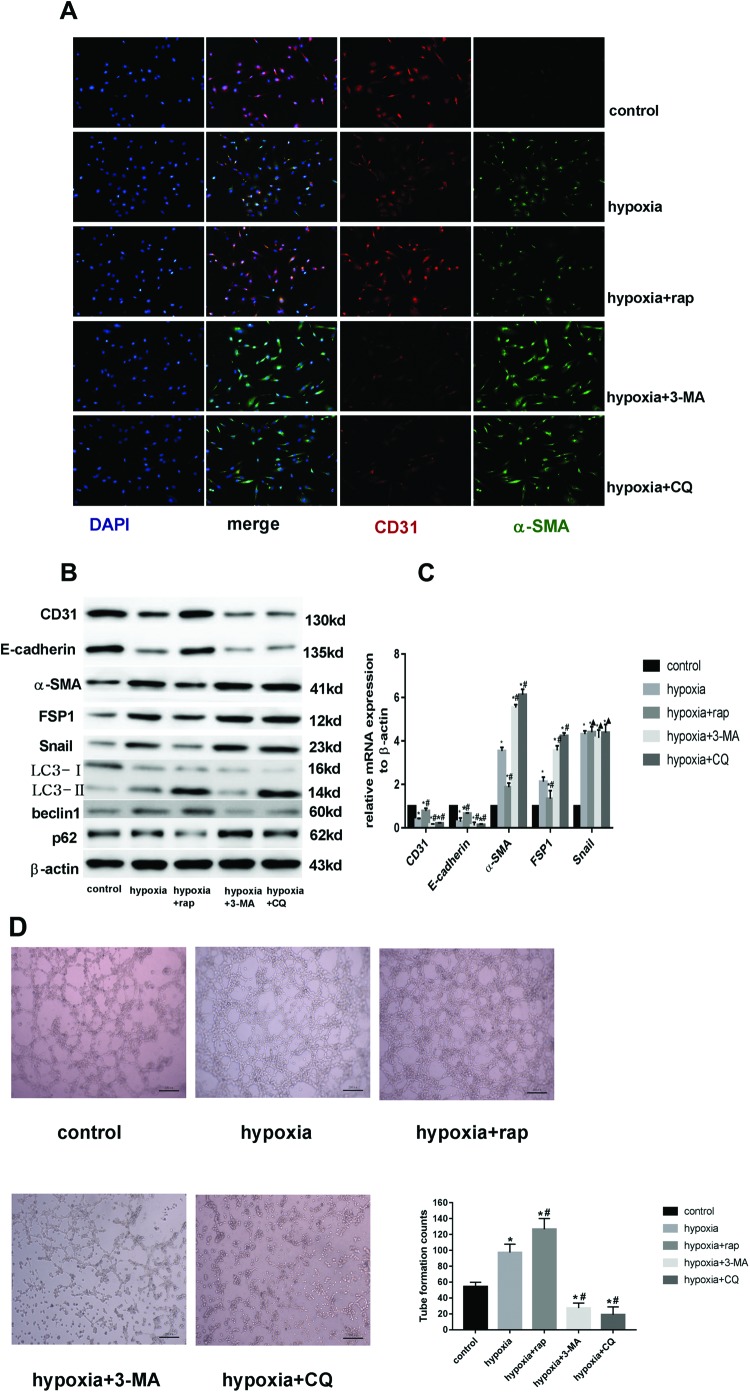
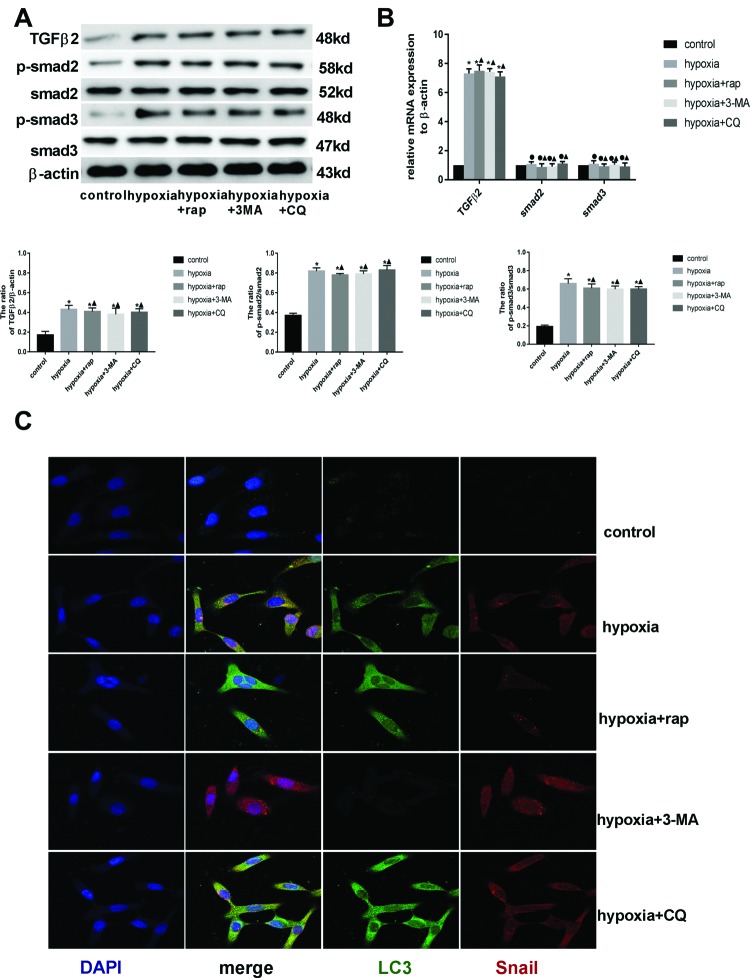
Endothelial-to-mesenchymal transition (EndMT) mainly exists in cardiovascular development and disease progression, and is well known to contribute to cardiac fibrosis. Recent studies indicated that autophagy also participates in the regulation of cardiac fibrosis. However, the precise role of autophagy in cardiac fibrosis and the underlying molecular mechanism remain unclear. The present study aimed to explore the role of autophagy in EndMT, reveal the underlying molecular mechanism, and seek new therapy for cardiac fibrosis. In the present study, we found that EndMT and autophagy were induced simultaneously by hypoxia in human cardiac microvascular endothelial cells (HCMECs). Rapamycin, an autophagy enhancer, attenuated EndMT with promoting angiogenesis, while 3-methyladenine (3-MA) and chloroquine (CQ), agents that inhibit autophagy, accelerated the progression accompanied by the decrease in counts of tube formation under hypoxia conditions. Interestingly, intervening autophagy by rapamycin, 3-MA, or CQ did not affect hypoxia-induced autocrine TGFβ signaling, but changed the expression of Snail protein without alterations in the expression of mRNA. Furthermore, the colocalization of LC3 and Snail indicated that autophagy might mediate Snail degradation under hypoxia conditions in HCMECs. Interaction of p62, the substrate of autophagy, with Snail by co-immunoprecipitation especially in hypoxia-incubated cells confirmed the hypothesis. In conclusion, autophagy serves as a cytoprotective mechanism against EndMT to promote angiogenesis by degrading Snail under hypoxia conditions, suggesting that autophagy targetted therapeutic strategies may be applicable for cardiac fibrosis by EndMT.
内皮细胞-间充质转化(EndMT)主要存在于心血管发育和疾病进展中,并且已知它有助于心脏纤维化。最近的研究表明,自噬也参与了心脏纤维化的调节。然而,自噬在心脏纤维化中的精确作用及其潜在的分子机制尚不清楚。本研究旨在探讨自噬在 EndMT 中的作用,揭示其潜在的分子机制,并为心脏纤维化寻求新的治疗方法。在本研究中,我们发现缺氧可同时诱导人心脏微血管内皮细胞(HCMECs)发生 EndMT 和自噬。自噬增强剂雷帕霉素通过促进血管生成来减轻 EndMT,而自噬抑制剂 3-甲基腺嘌呤(3-MA)和氯喹(CQ)在缺氧条件下加速了 EndMT 的进展,同时伴随着管形成计数的减少。有趣的是,雷帕霉素、3-MA 或 CQ 干预自噬并不影响缺氧诱导的自分泌 TGFβ信号,但改变了 Snail 蛋白的表达,而不改变 mRNA 的表达。此外,LC3 和 Snail 的共定位表明,自噬可能在缺氧条件下通过降解 HCMECs 中的 Snail 来介导 Snail 降解。共免疫沉淀中 p62(自噬的底物)与 Snail 的相互作用,特别是在缺氧孵育的细胞中,证实了这一假设。总之,自噬作为一种细胞保护机制,通过在缺氧条件下降解 Snail 来促进血管生成,从而抑制 EndMT,提示针对自噬的治疗策略可能适用于通过 EndMT 导致的心脏纤维化。