Gorfine Malka, Berndt Sonja I, Chang-Claude Jenny, Hoffmeister Michael, Le Marchand Loic, Potter John, Slattery Martha L, Keret Nir, Peters Ulrike, Hsu Li
Department of Statistics and Operations Research, Tel Aviv University, Tel Aviv, Israel.
Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health, Bethesda, Maryland, United States of America.
PLoS One. 2017 Aug 16;12(8):e0181269. doi: 10.1371/journal.pone.0181269. eCollection 2017.
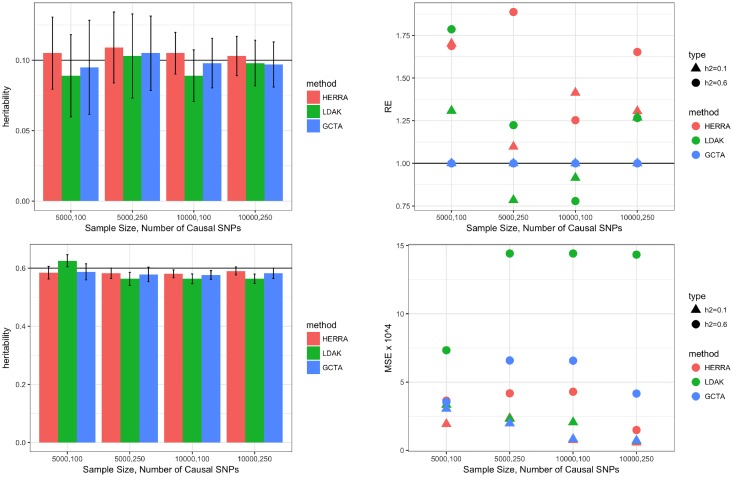
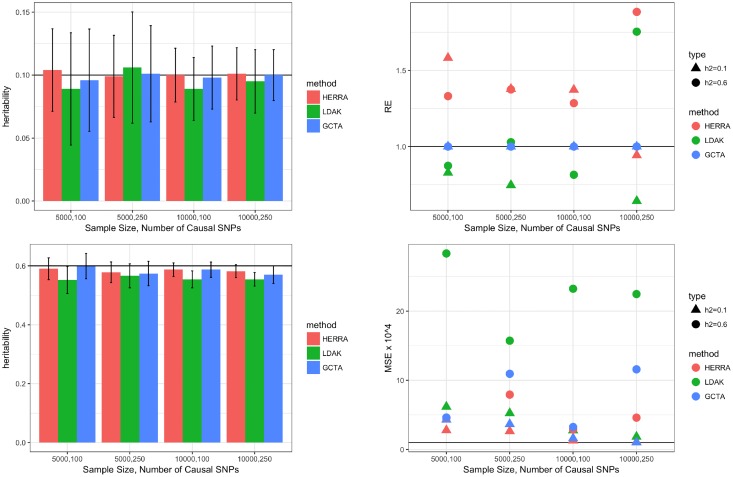
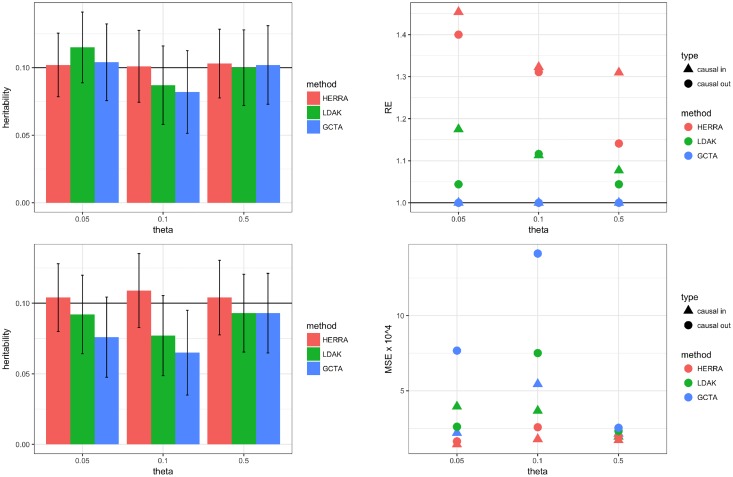
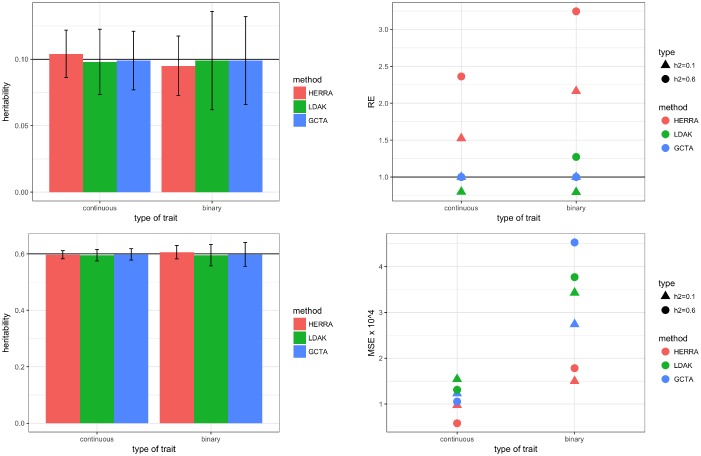
The popular Genome-wide Complex Trait Analysis (GCTA) software uses the random-effects models for estimating the narrow-sense heritability based on GWAS data of unrelated individuals without knowing and identifying the causal loci. Many methods have since extended this approach to various situations. However, since the proportion of causal loci among the variants is typically very small and GCTA uses all variants to calculate the similarities among individuals, the estimation of heritability may be unstable, resulting in a large variance of the estimates. Moreover, if the causal SNPs are not genotyped, GCTA sometimes greatly underestimates the true heritability. We present a novel narrow-sense heritability estimator, named HERRA, using well-developed ultra-high dimensional machine-learning methods, applicable to continuous or dichotomous outcomes, as other existing methods. Additionally, HERRA is applicable to time-to-event or age-at-onset outcome, which, to our knowledge, no existing method can handle. Compared to GCTA and LDAK for continuous and binary outcomes, HERRA often has a smaller variance, and when causal SNPs are not genotyped, HERRA has a much smaller empirical bias. We applied GCTA, LDAK and HERRA to a large colorectal cancer dataset using dichotomous outcome (4,312 cases, 4,356 controls, genotyped using Illumina 300K), the respective heritability estimates of GCTA, LDAK and HERRA are 0.068 (SE = 0.017), 0.072 (SE = 0.021) and 0.110 (SE = 5.19 x 10-3). HERRA yields over 50% increase in heritability estimate compared to GCTA or LDAK.
广受欢迎的全基因组复杂性状分析(GCTA)软件使用随机效应模型,基于无关个体的全基因组关联研究(GWAS)数据来估计狭义遗传力,而无需知晓和识别因果位点。此后,许多方法将这种方法扩展到了各种情况。然而,由于变异中因果位点的比例通常非常小,且GCTA使用所有变异来计算个体间的相似性,遗传力估计可能不稳定,导致估计值的方差很大。此外,如果因果单核苷酸多态性(SNP)未进行基因分型,GCTA有时会大大低估真实的遗传力。我们提出了一种新的狭义遗传力估计方法,名为HERRA,它使用了成熟的超高维机器学习方法,与其他现有方法一样,适用于连续或二分结局。此外,HERRA适用于事件发生时间或发病年龄结局,据我们所知,现有方法无法处理此类情况。与用于连续和二元结局的GCTA和LDAK相比,HERRA的方差通常较小,并且当因果SNP未进行基因分型时,HERRA的经验偏差要小得多。我们将GCTA、LDAK和HERRA应用于一个大型结直肠癌数据集,使用二分结局(4312例病例,4356例对照,使用Illumina 300K进行基因分型),GCTA、LDAK和HERRA各自的遗传力估计值分别为0.068(标准误=0.017)、0.072(标准误=0.021)和0.110(标准误=5.19×10⁻³)。与GCTA或LDAK相比,HERRA的遗传力估计值提高了50%以上。