Biomedical Informatics, Research Branch, Sidra Medical and Research Center, Post Box No. 26999, Doha, Qatar.
Sci Rep. 2017 Aug 22;7(1):9058. doi: 10.1038/s41598-017-09089-1.
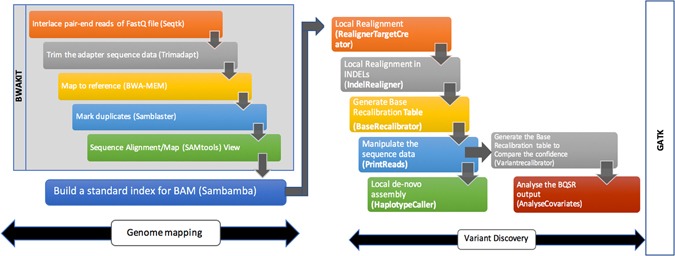
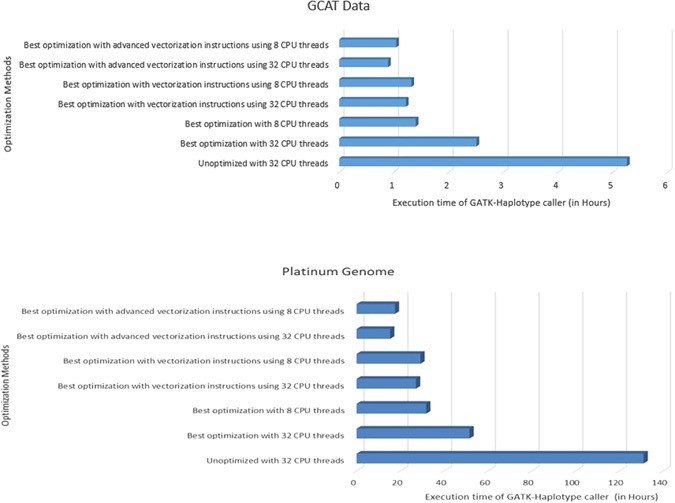
Next generation sequencing (NGS) data analysis is highly compute intensive. In-memory computing, vectorization, bulk data transfer, CPU frequency scaling are some of the hardware features in the modern computing architectures. To get the best execution time and utilize these hardware features, it is necessary to tune the system level parameters before running the application. We studied the GATK-HaplotypeCaller which is part of common NGS workflows, that consume more than 43% of the total execution time. Multiple GATK 3.x versions were benchmarked and the execution time of HaplotypeCaller was optimized by various system level parameters which included: (i) tuning the parallel garbage collection and kernel shared memory to simulate in-memory computing, (ii) architecture-specific tuning in the PairHMM library for vectorization, (iii) including Java 1.8 features through GATK source code compilation and building a runtime environment for parallel sorting and bulk data transfer (iv) the default 'on-demand' mode of CPU frequency is over-clocked by using 'performance-mode' to accelerate the Java multi-threads. As a result, the HaplotypeCaller execution time was reduced by 82.66% in GATK 3.3 and 42.61% in GATK 3.7. Overall, the execution time of NGS pipeline was reduced to 70.60% and 34.14% for GATK 3.3 and GATK 3.7 respectively.
下一代测序 (NGS) 数据分析计算量很大。现代计算架构中的一些硬件特性包括内存计算、向量化、批量数据传输、CPU 频率调整等。为了获得最佳的执行时间并利用这些硬件特性,在运行应用程序之前,有必要调整系统级参数。我们研究了 GATK-HaplotypeCaller,它是常见 NGS 工作流程的一部分,占总执行时间的 43%以上。我们对多个 GATK 3.x 版本进行了基准测试,并通过各种系统级参数优化了 HaplotypeCaller 的执行时间,其中包括:(i) 调整并行垃圾收集和内核共享内存以模拟内存计算,(ii) 在 PairHMM 库中进行特定于架构的调整以实现向量化,(iii) 通过 GATK 源代码编译和构建并行排序和批量数据传输的运行时环境来包含 Java 1.8 特性,(iv) 使用“性能模式”将 CPU 频率的默认“按需”模式超频,以加速 Java 多线程。结果,在 GATK 3.3 中,HaplotypeCaller 的执行时间减少了 82.66%,在 GATK 3.7 中减少了 42.61%。总体而言,NGS 管道的执行时间分别减少了 70.60%和 34.14%,适用于 GATK 3.3 和 GATK 3.7。