Lee Sei-Jung, Jung Young Hyun, Kim Jun Sung, Lee Hyun Jik, Lee Sang Hun, Lee Kyu-Ho, Jang Kyung Ku, Choi Sang Ho, Han Ho Jae
Department of Pharmaceutical Engineering, Daegu Haany UniversityGyeongsan, South Korea.
Department of Veterinary Physiology, College of Veterinary Medicine, Research Institute for Veterinary Science and BK21 PLUS Program for Creative Veterinary Science Research Center, Seoul National UniversitySeoul, South Korea.
Front Cell Infect Microbiol. 2017 Aug 11;7:352. doi: 10.3389/fcimb.2017.00352. eCollection 2017.
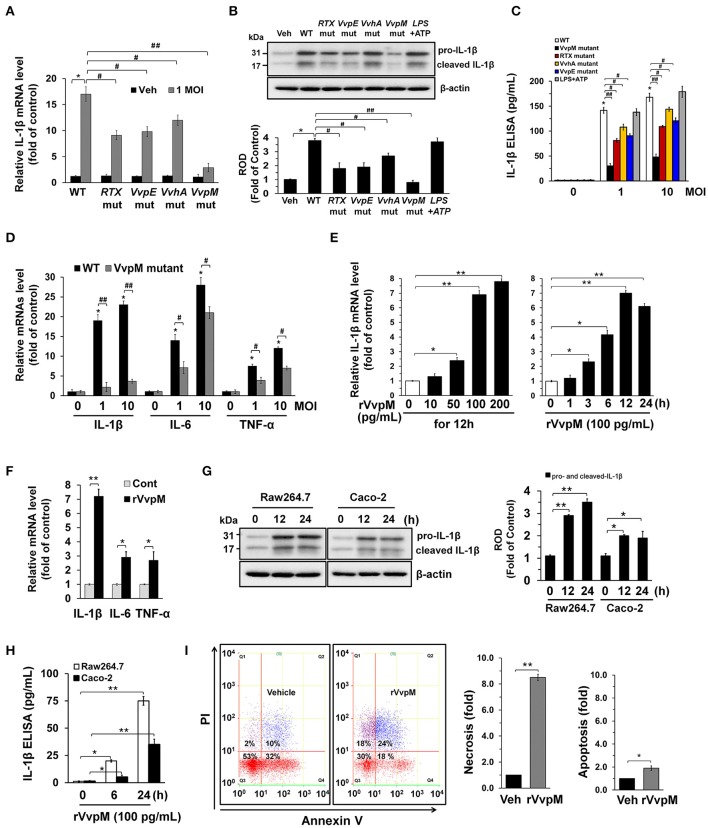
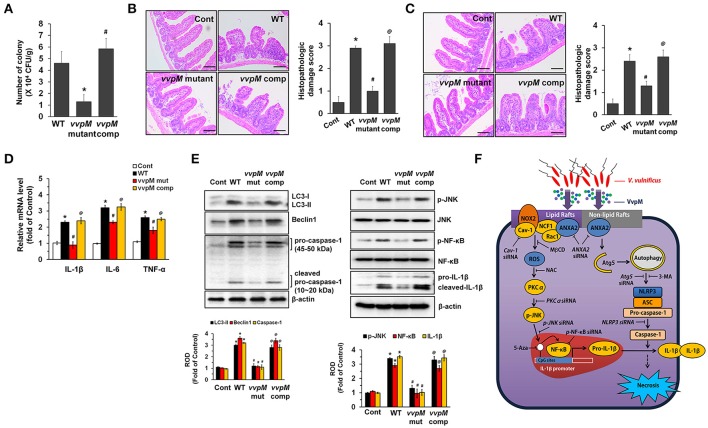
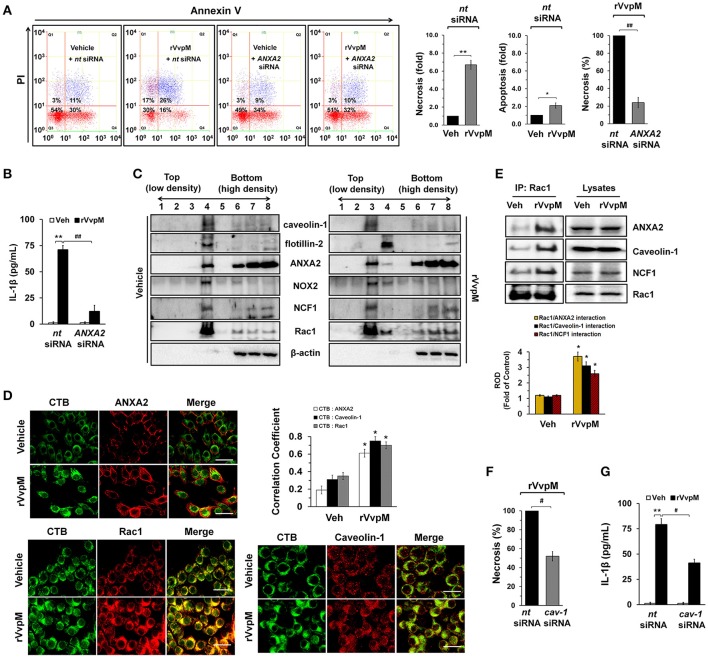
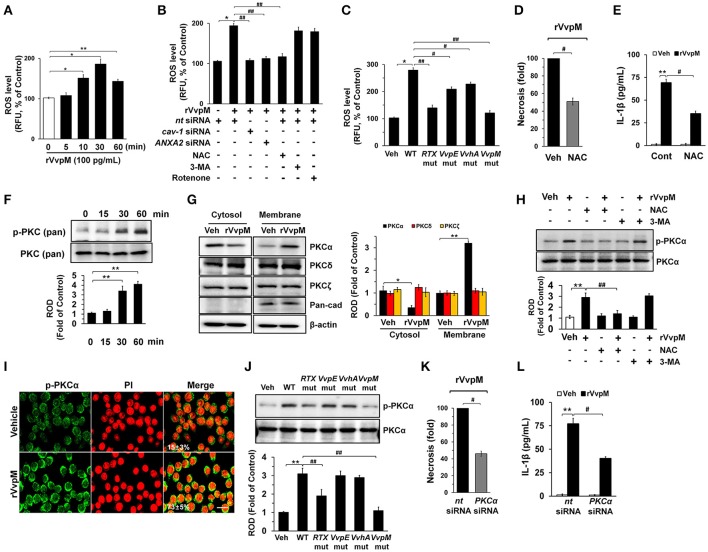
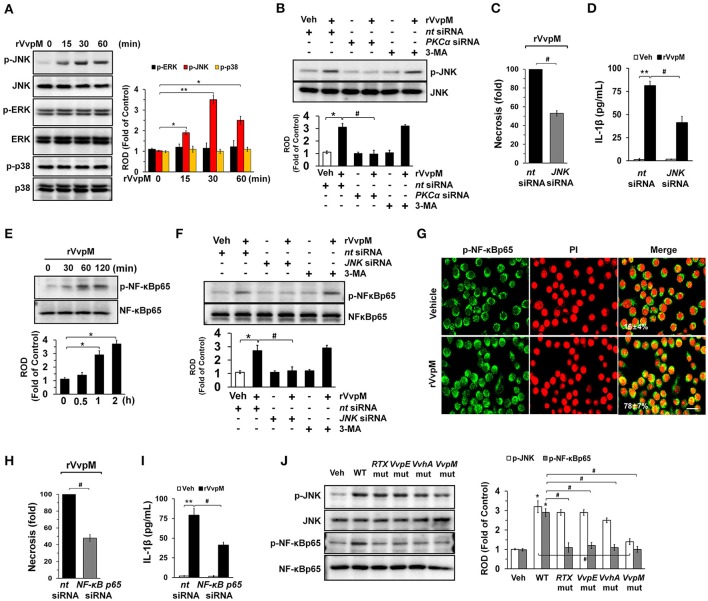
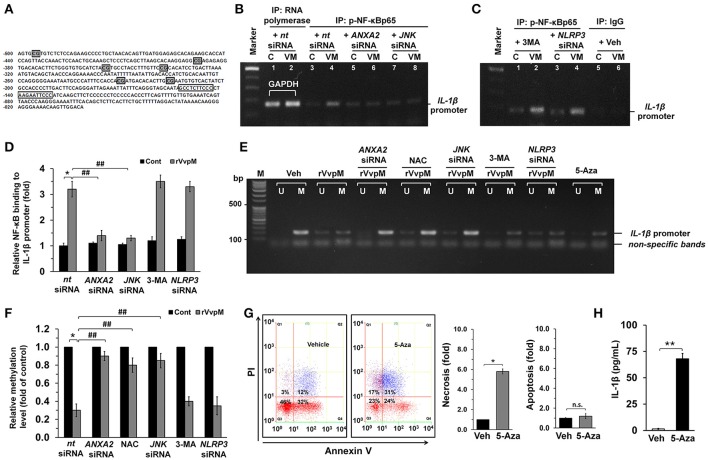
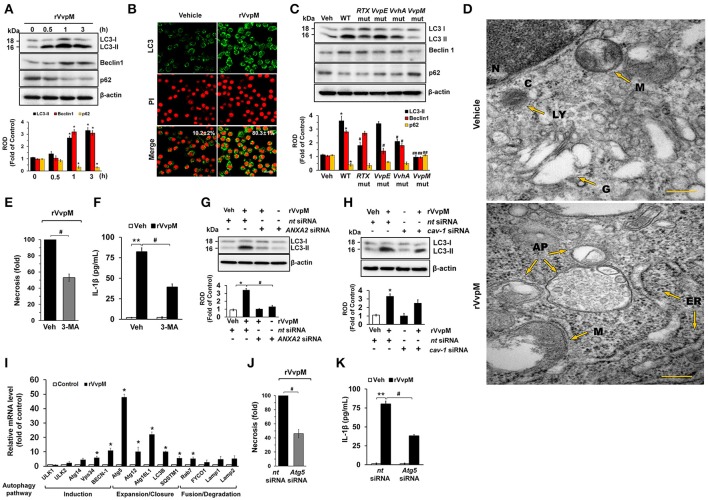
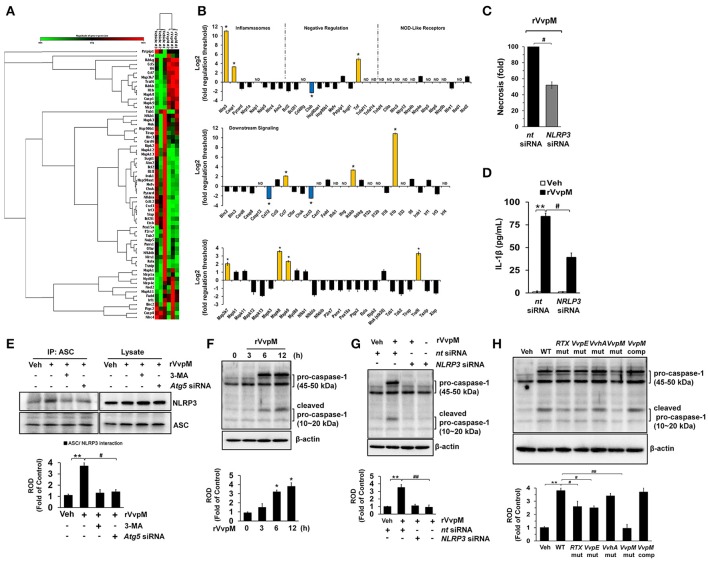
An inflammatory form of phagocyte death evoked by the Gram-negative bacterium (WT) is one of hallmarks to promote their colonization, but the virulence factor and infectious mechanism involved in this process remain largely unknown. Here, we identified extracellular metalloprotease VvpM as a new virulence factor and investigated the molecular mechanism of VvpM which acts during the regulation of the inflammatory form of macrophage death and bacterial colonization. Mutation of the gene appeared to play major role in the prevention of IL-1β production due to infection in macrophage. However, the recombinant protein (r) VvpM caused IL-1β production coupled with necrotic cell death, which is highly susceptible to the knockdown of annexin A2 (ANXA2) located in both membrane lipid and non-lipid rafts. In lipid rafts, rVvpM recruited NOX enzymes coupled with ANXA2 to facilitate the production of ROS responsible for the epigenetic and transcriptional regulation of NF-κB in the IL-1β promoter. rVvpM acting on non-lipid rafts increased LC3 puncta formation and autophagic flux, which are required for the mRNA expression of involved in the autophagosome formation process. The autophagy activation caused by rVvpM induced NLRP3 inflammasome-dependent caspase-1 activation in the promoting of IL-1β production. In mouse models of infection, the mutant failed to elevate the level of pro-inflammatory responses closely related to IL-1β production and prevented bacterial colonization. These findings delineate efficiently regulates two pathogenic pathways that stimulate NF-κB-dependent IL-1β production and autophagy-mediated NLRP3 inflammasome via distinct spatial targeting by ANXA2.
由革兰氏阴性菌(野生型)引发的一种炎症性吞噬细胞死亡形式是促进其定植的标志之一,但该过程中涉及的毒力因子和感染机制在很大程度上仍不清楚。在此,我们鉴定出细胞外金属蛋白酶VvpM为一种新的毒力因子,并研究了VvpM在调节巨噬细胞炎症性死亡和细菌定植过程中发挥作用的分子机制。基因的突变似乎在预防巨噬细胞因感染而产生白细胞介素-1β中起主要作用。然而,重组蛋白(r)VvpM导致白细胞介素-1β产生并伴有坏死性细胞死亡,这对位于膜脂筏和非脂筏中的膜联蛋白A2(ANXA2)的敲低高度敏感。在脂筏中,rVvpM募集与ANXA2结合的NADPH氧化酶,以促进活性氧的产生,活性氧负责白细胞介素-1β启动子中核因子-κB的表观遗传和转录调控。作用于非脂筏的rVvpM增加了LC3斑点的形成和自噬通量,这是自噬体形成过程中相关基因mRNA表达所必需的。rVvpM引起的自噬激活在促进白细胞介素-1β产生过程中诱导了NLRP3炎性小体依赖性半胱天冬酶-1的激活。在感染的小鼠模型中,突变体未能提高与白细胞介素-1β产生密切相关的促炎反应水平,并阻止了细菌定植。这些发现表明,VvpM通过ANXA2的不同空间靶向有效地调节了两条致病途径,即刺激核因子-κB依赖性白细胞介素-1β产生和自噬介导的NLRP3炎性小体。