Department for Anaesthesiology, Heinrich Heine University, 40225 Düsseldorf, Germany.
BMFZ, Heinrich Heine University, 40225 Düsseldorf, Germany.
Int J Mol Sci. 2017 Sep 5;18(9):1900. doi: 10.3390/ijms18091900.
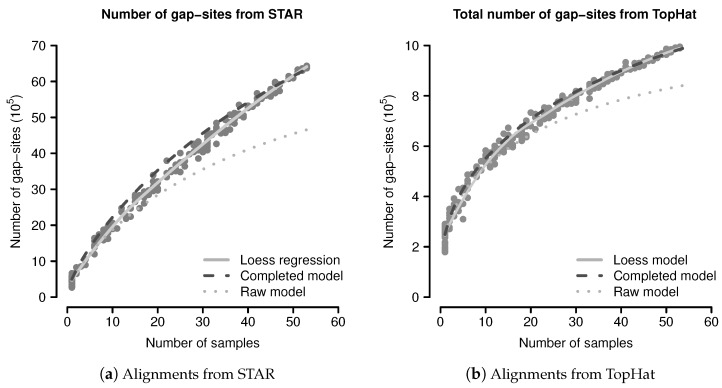
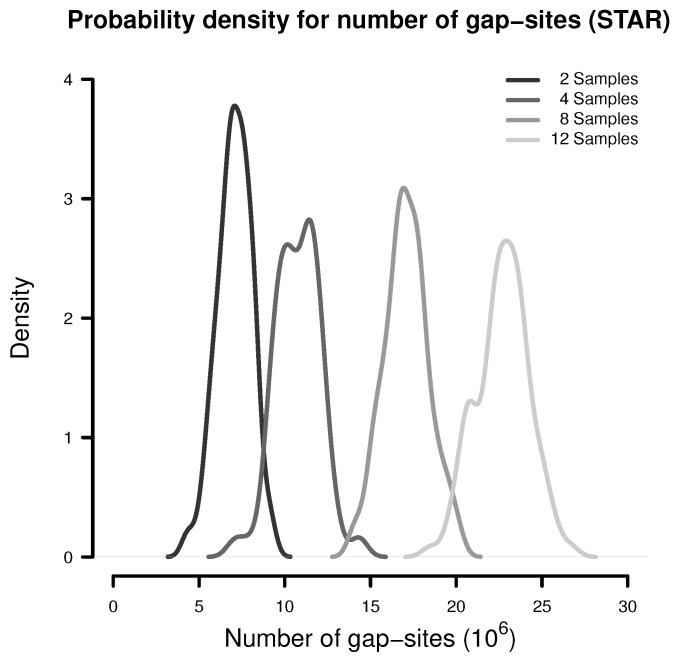
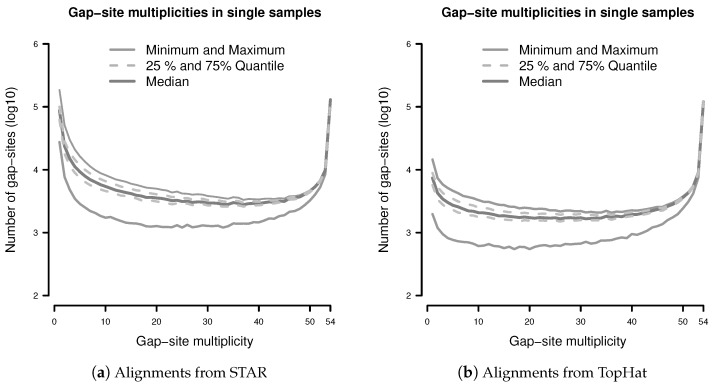
Merging data from multiple samples is required to detect low expressed transcripts or splicing events that might be present only in a subset of samples. However, the exact number of required replicates enabling the detection of such rare events often remains a mystery but can be approached through probability theory. Here, we describe a probabilistic model, relating the number of observed events in a batch of samples with observation probabilities. Therein, samples appear as a heterogeneous collection of events, which are observed with some probability. The model is evaluated in a batch of 54 transcriptomes of human dermal fibroblast samples. The majority of putative splice-sites (alignment gap-sites) are detected in (almost) all samples or only sporadically, resulting in an U-shaped pattern for observation probabilities. The probabilistic model systematically underestimates event numbers due to a bias resulting from finite sampling. However, using an additional assumption, the probabilistic model can predict observed event numbers within a <10% deviation from the median. Single samples contain a considerable amount of uniquely observed putative splicing events (mean 7122 in alignments from TopHat alignments and 86,215 in alignments from STAR). We conclude that the probabilistic model provides an adequate description for observation of gap-sites in transcriptome data. Thus, the calculation of required sample sizes can be done by application of a simple binomial model to sporadically observed random events. Due to the large number of uniquely observed putative splice-sites and the known stochastic noise in the splicing machinery, it appears advisable to include observation of rare splicing events into analysis objectives. Therefore, it is beneficial to take scores for the validation of gap-sites into account.
合并来自多个样本的数据是检测低表达转录本或剪接事件所必需的,这些事件可能只存在于一部分样本中。然而,能够检测到这些罕见事件的确切重复次数通常仍然是个谜,但可以通过概率论来接近。在这里,我们描述了一种概率模型,将一批样本中观察到的事件数量与观察概率联系起来。在该模型中,样本被视为事件的异质集合,这些事件以一定的概率被观察到。该模型在 54 个人类真皮成纤维细胞样本的转录组中进行了评估。大多数假定的剪接位点(比对缺口位点)在(几乎)所有样本中或仅偶尔被检测到,导致观察概率呈 U 形。由于有限采样导致的偏差,概率模型系统地低估了事件数量。然而,通过使用额外的假设,概率模型可以预测观察到的事件数量,其与中位数的偏差小于 10%。单个样本包含相当数量的独特观察到的假定剪接事件(来自 TopHat 比对的比对中的平均值为 7122 个,来自 STAR 的比对中的平均值为 86215 个)。我们得出结论,概率模型为观察转录组数据中的缺口位点提供了一个充分的描述。因此,可以通过应用简单的二项式模型来计算偶尔观察到的随机事件所需的样本量。由于独特观察到的假定剪接事件的数量很大,以及剪接机制中已知的随机噪声,似乎有必要将稀有剪接事件的观察纳入分析目标。因此,考虑缺口位点的分数以验证其是有益的。