Sun Shiquan, Hood Michelle, Scott Laura, Peng Qinke, Mukherjee Sayan, Tung Jenny, Zhou Xiang
Systems Engineering Institute, Xi'an Jiaotong University, Xi'an, Shaanxi 710049, P.R. China.
Department of Biostatistics, University of Michigan, Ann Arbor, MI 48109, USA.
Nucleic Acids Res. 2017 Jun 20;45(11):e106. doi: 10.1093/nar/gkx204.
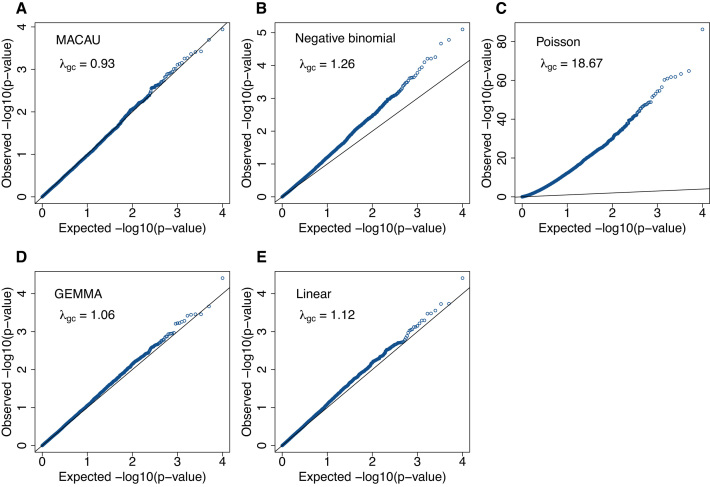
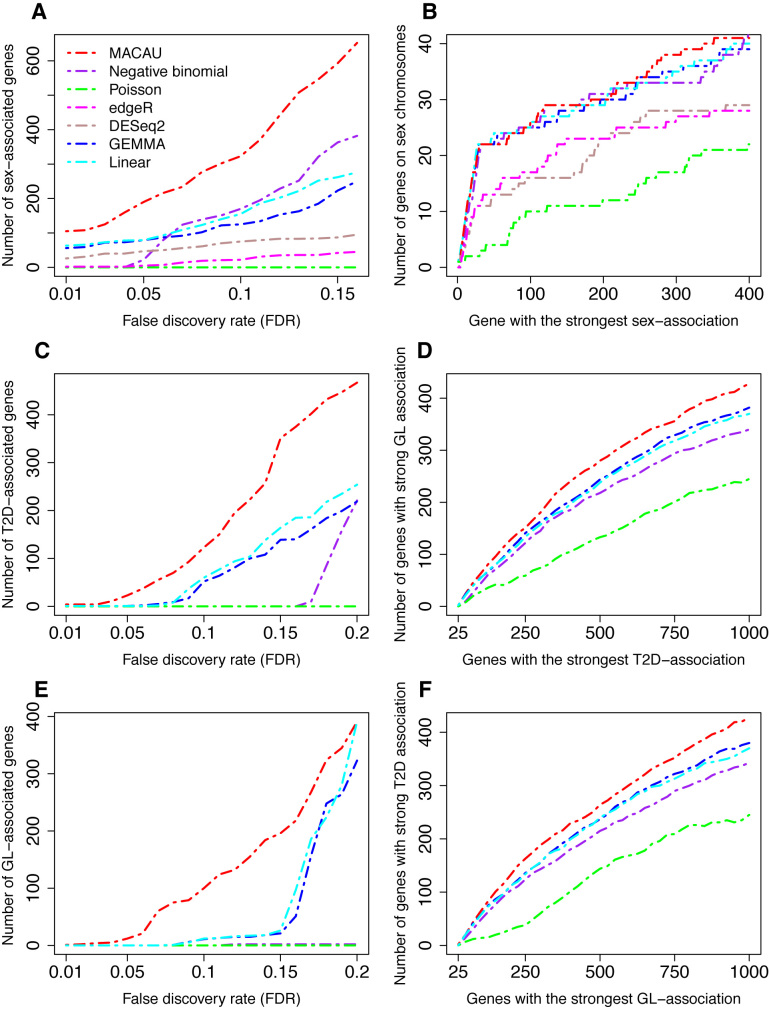
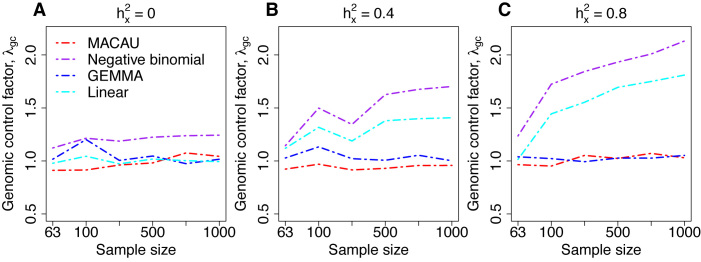
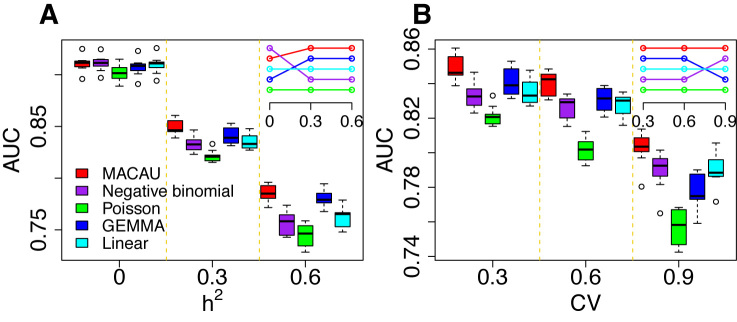
Identifying differentially expressed (DE) genes from RNA sequencing (RNAseq) studies is among the most common analyses in genomics. However, RNAseq DE analysis presents several statistical and computational challenges, including over-dispersed read counts and, in some settings, sample non-independence. Previous count-based methods rely on simple hierarchical Poisson models (e.g. negative binomial) to model independent over-dispersion, but do not account for sample non-independence due to relatedness, population structure and/or hidden confounders. Here, we present a Poisson mixed model with two random effects terms that account for both independent over-dispersion and sample non-independence. We also develop a scalable sampling-based inference algorithm using a latent variable representation of the Poisson distribution. With simulations, we show that our method properly controls for type I error and is generally more powerful than other widely used approaches, except in small samples (n <15) with other unfavorable properties (e.g. small effect sizes). We also apply our method to three real datasets that contain related individuals, population stratification or hidden confounders. Our results show that our method increases power in all three data compared to other approaches, though the power gain is smallest in the smallest sample (n = 6). Our method is implemented in MACAU, freely available at www.xzlab.org/software.html.
从RNA测序(RNAseq)研究中识别差异表达(DE)基因是基因组学中最常见的分析之一。然而,RNAseq差异表达分析存在一些统计和计算挑战,包括过度分散的读数计数,以及在某些情况下样本的非独立性。以前基于计数的方法依赖于简单的分层泊松模型(如负二项分布)来对独立的过度分散进行建模,但没有考虑由于相关性、群体结构和/或隐藏的混杂因素导致的样本非独立性。在这里,我们提出了一个具有两个随机效应项的泊松混合模型,该模型同时考虑了独立的过度分散和样本非独立性。我们还使用泊松分布的潜在变量表示开发了一种基于采样的可扩展推理算法。通过模拟,我们表明我们的方法能够正确控制I型错误,并且除了在具有其他不利特性(如小效应量)的小样本(n <15)中,通常比其他广泛使用的方法更强大。我们还将我们的方法应用于三个包含相关个体、群体分层或隐藏混杂因素的真实数据集。我们的结果表明,与其他方法相比,我们的方法在所有三个数据集中都提高了检验效能,尽管在最小的样本(n = 6)中效能增益最小。我们的方法在MACAU中实现,可在www.xzlab.org/software.html上免费获取。