Feng Shuang, Bowden Neil, Fragiadaki Maria, Souilhol Celine, Hsiao Sarah, Mahmoud Marwa, Allen Scott, Pirri Daniela, Ayllon Blanca Tardajos, Akhtar Shamima, Thompson A A Roger, Jo Hanjoong, Weber Christian, Ridger Victoria, Schober Andreas, Evans Paul C
From the Department of Infection, Immunity, and Cardiovascular Disease, INSIGNEO Institute for In Silico Medicine, and the Bateson Centre (S.F., N.B., M.F., C.S., H.S., M.M., D.P., B.T.A., A.A.R.T., V.R., P.C.E.) and Sheffield Institute for Translational Neuroscience (S.A.), University of Sheffield, United Kingdom; Institute for Cardiovascular Prevention, Ludwig-Maximilians University of Munich and DZHK (German Centre for Cardiovascular Research), partner site Munich Heart Alliance, Germany (S.A., C.W., A.S.); and Wallace H. Coulter Department of Biomedical Engineering, Georgia Institute of Technology and Emory University, Atlanta (H.J.).
Arterioscler Thromb Vasc Biol. 2017 Nov;37(11):2087-2101. doi: 10.1161/ATVBAHA.117.309249. Epub 2017 Sep 7.
Atherosclerosis develops near branches and bends of arteries that are exposed to low shear stress (mechanical drag). These sites are characterized by excessive endothelial cell (EC) proliferation and inflammation that promote lesion initiation. The transcription factor HIF1α (hypoxia-inducible factor 1α) is canonically activated by hypoxia and has a role in plaque neovascularization. We studied the influence of shear stress on HIF1α activation and the contribution of this noncanonical pathway to lesion initiation.
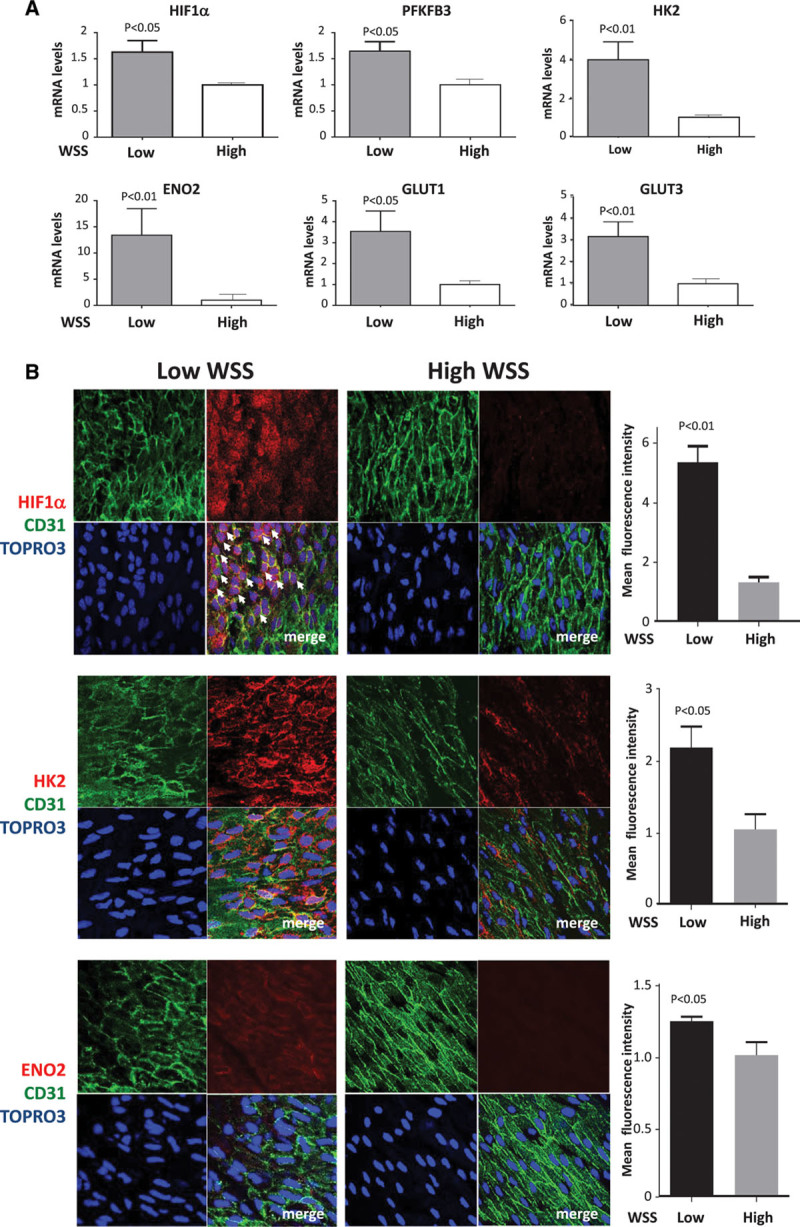
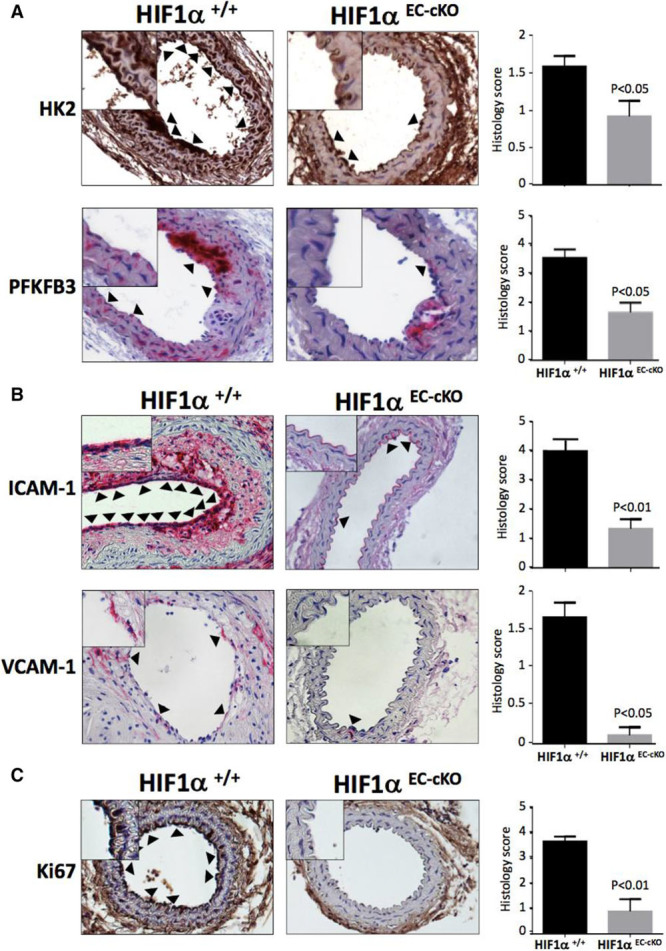
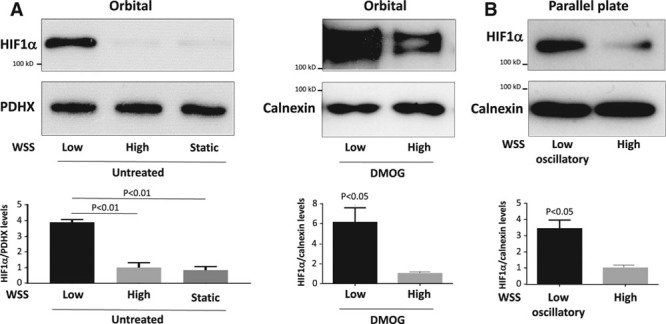
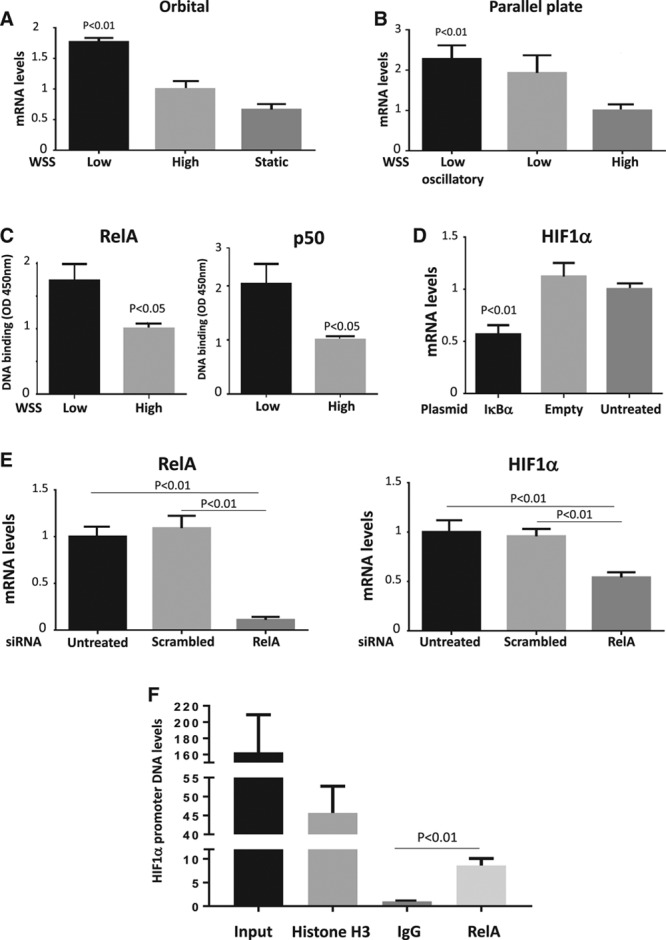
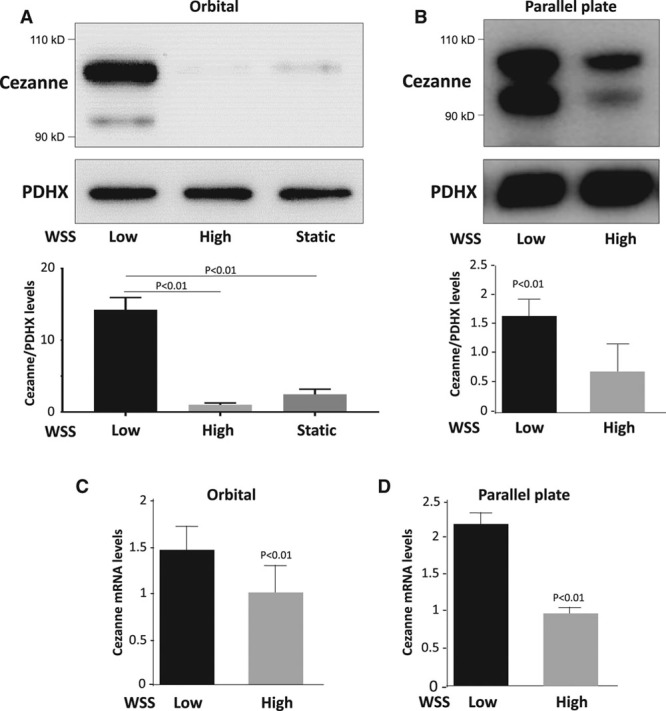
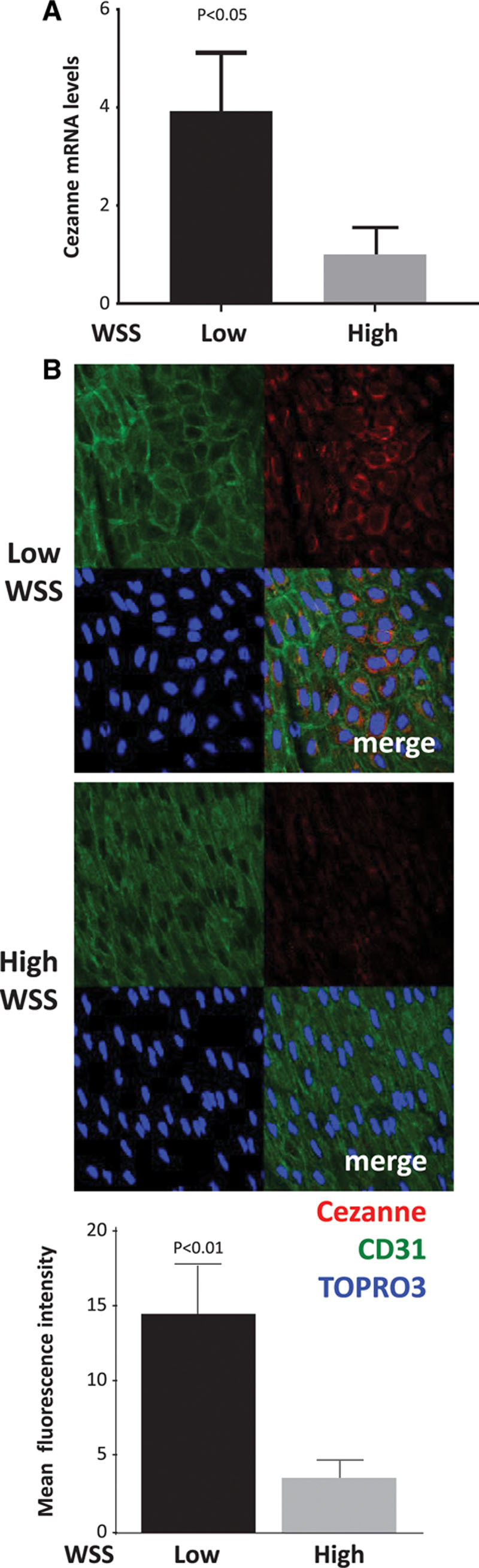
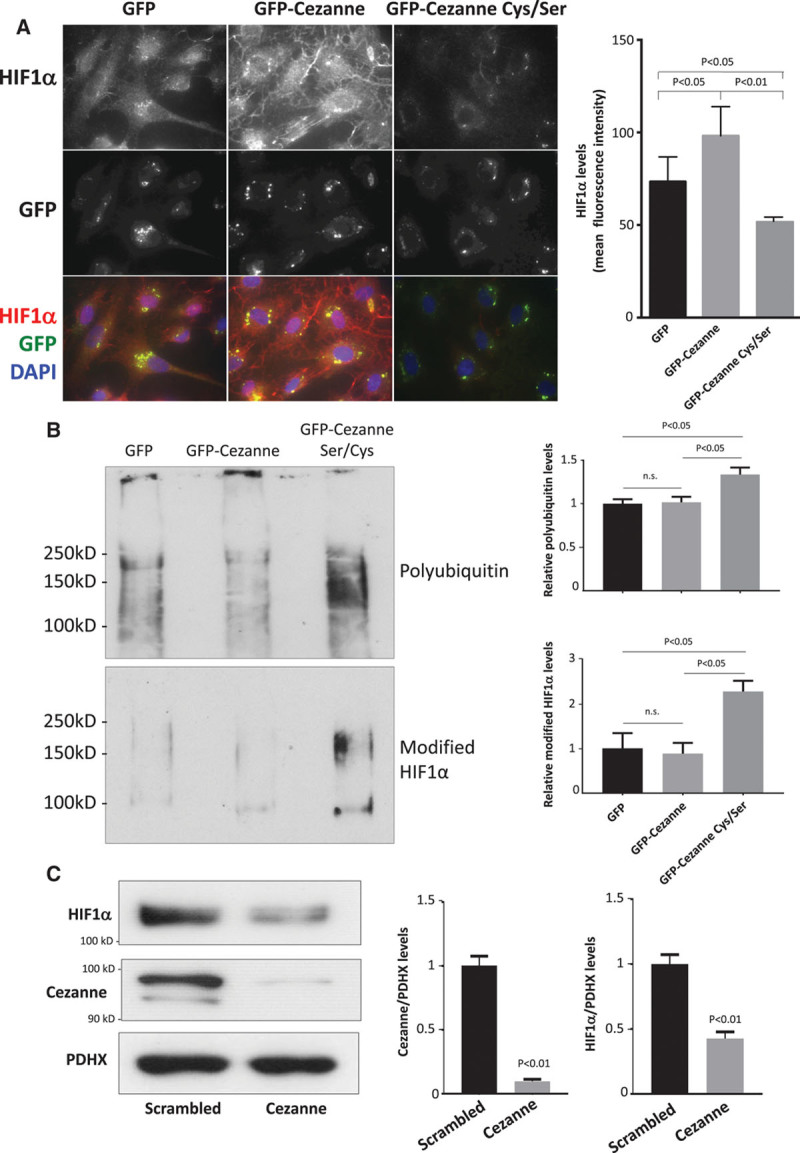
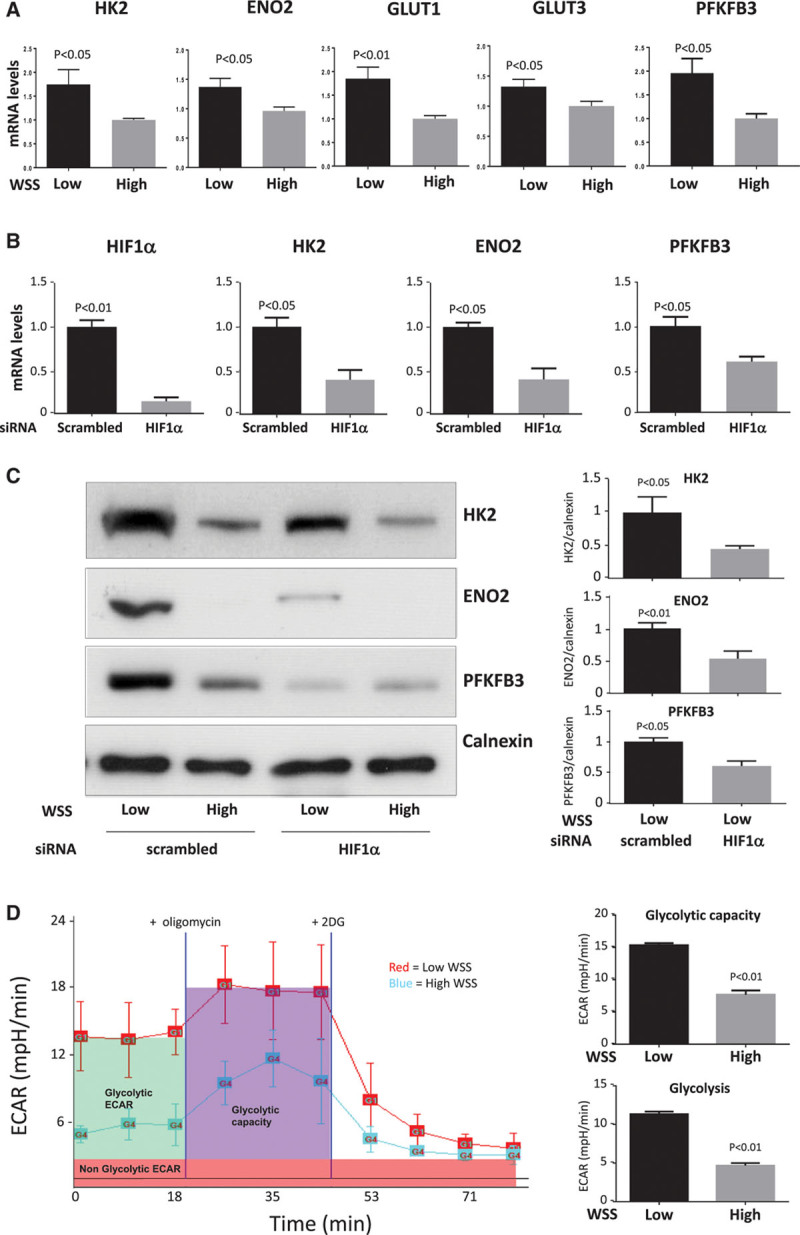
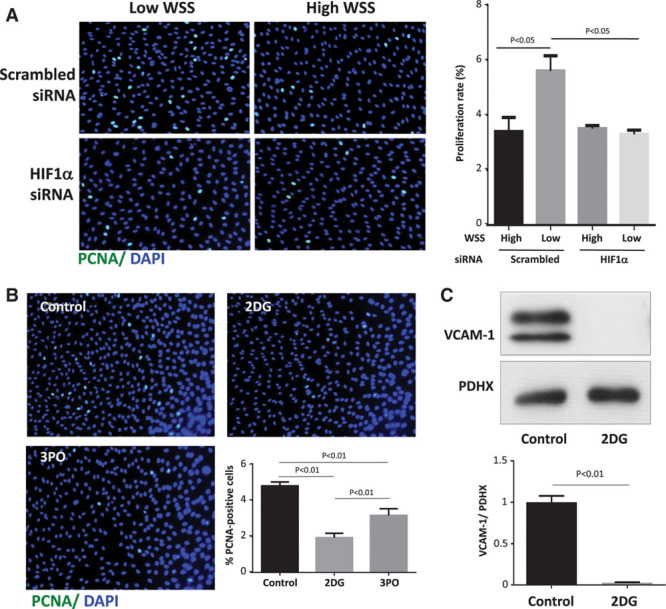
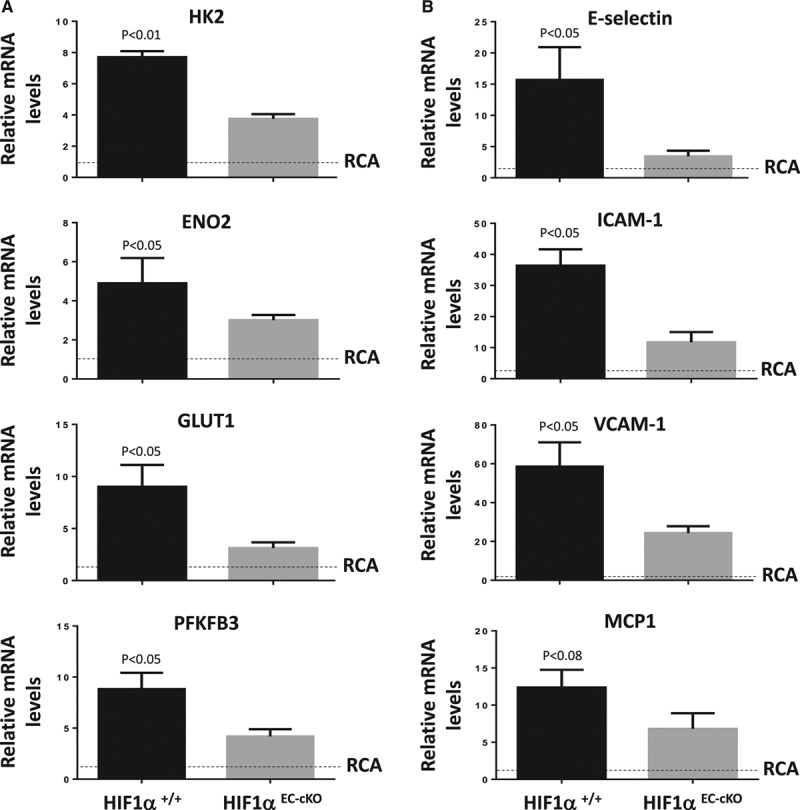
Quantitative polymerase chain reaction and en face staining revealed that HIF1α was expressed preferentially at low shear stress regions of porcine and murine arteries. Low shear stress induced HIF1α in cultured EC in the presence of atmospheric oxygen. The mechanism involves the transcription factor nuclear factor-κB that induced HIF1α transcripts and induction of the deubiquitinating enzyme Cezanne that stabilized HIF1α protein. Gene silencing revealed that HIF1α enhanced proliferation and inflammatory activation in EC exposed to low shear stress via induction of glycolysis enzymes. We validated this observation by imposing low shear stress in murine carotid arteries (partial ligation) that upregulated the expression of HIF1α, glycolysis enzymes, and inflammatory genes and enhanced EC proliferation. EC-specific genetic deletion of HIF1α in hypercholesterolemic apolipoprotein E-defecient mice reduced inflammation and endothelial proliferation in partially ligated arteries, indicating that HIF1α drives inflammation and vascular dysfunction at low shear stress regions.
Mechanical low shear stress activates HIF1α at atheroprone regions of arteries via nuclear factor-κB and Cezanne. HIF1α promotes atherosclerosis initiation at these sites by inducing excessive EC proliferation and inflammation via the induction of glycolysis enzymes.
动脉粥样硬化在暴露于低剪切应力(机械阻力)的动脉分支和弯曲处附近发展。这些部位的特征是内皮细胞(EC)过度增殖和炎症,促进病变起始。转录因子HIF1α(缺氧诱导因子1α)通常由缺氧激活,并在斑块新生血管形成中起作用。我们研究了剪切应力对HIF1α激活的影响以及这种非经典途径对病变起始的贡献。
定量聚合酶链反应和整体染色显示,HIF1α在猪和鼠动脉的低剪切应力区域优先表达。在大气氧存在的情况下,低剪切应力在培养的内皮细胞中诱导HIF1α。其机制涉及诱导HIF1α转录本的转录因子核因子-κB以及稳定HIF1α蛋白的去泛素化酶塞尚的诱导。基因沉默显示,HIF1α通过诱导糖酵解酶增强了暴露于低剪切应力的内皮细胞的增殖和炎症激活。我们通过对鼠颈动脉施加低剪切应力(部分结扎)来验证这一观察结果,该操作上调了HIF1α、糖酵解酶和炎症基因的表达,并增强了内皮细胞增殖。在高胆固醇血症载脂蛋白E缺陷小鼠中,内皮细胞特异性基因缺失HIF1α可减少部分结扎动脉中的炎症和内皮细胞增殖,表明HIF1α在低剪切应力区域驱动炎症和血管功能障碍。
机械性低剪切应力通过核因子-κB和塞尚在动脉易患动脉粥样硬化的区域激活HIF1α。HIF1α通过诱导糖酵解酶诱导内皮细胞过度增殖和炎症,从而促进这些部位的动脉粥样硬化起始。