Division of Molecular Pathology, The Institute of Cancer Research, London, United Kingdom.
Centre for Molecular Pathology, Royal Marsden Hospital, London, United Kingdom.
Sci Rep. 2017 Sep 7;7(1):10849. doi: 10.1038/s41598-017-11110-6.
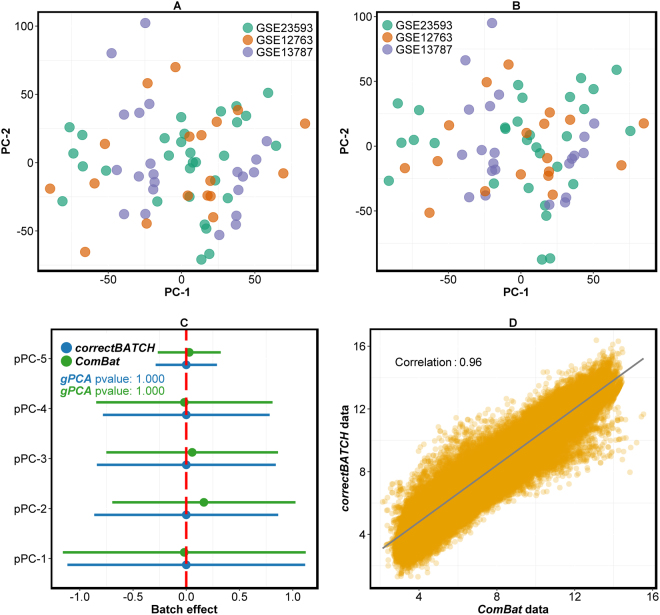
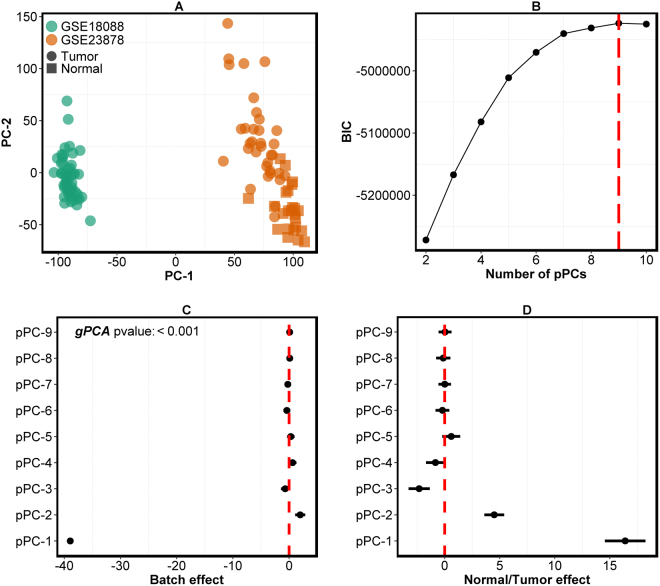
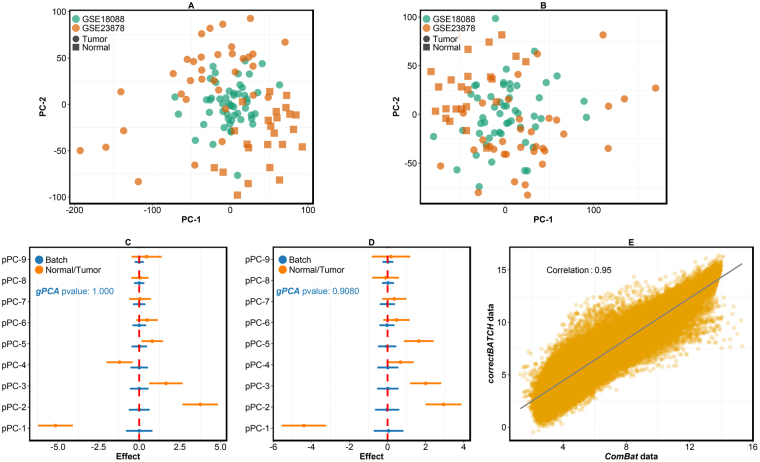
Genome projects now generate large-scale data often produced at various time points by different laboratories using multiple platforms. This increases the potential for batch effects. Currently there are several batch evaluation methods like principal component analysis (PCA; mostly based on visual inspection), and sometimes they fail to reveal all of the underlying batch effects. These methods can also lead to the risk of unintentionally correcting biologically interesting factors attributed to batch effects. Here we propose a novel statistical method, finding batch effect (findBATCH), to evaluate batch effect based on probabilistic principal component and covariates analysis (PPCCA). The same framework also provides a new approach to batch correction, correcting batch effect (correctBATCH), which we have shown to be a better approach to traditional PCA-based correction. We demonstrate the utility of these methods using two different examples (breast and colorectal cancers) by merging gene expression data from different studies after diagnosing and correcting for batch effects and retaining the biological effects. These methods, along with conventional visual inspection-based PCA, are available as a part of an R package exploring batch effect (exploBATCH; https://github.com/syspremed/exploBATCH ).
基因组项目现在生成大规模数据,这些数据通常由不同实验室在不同时间点使用多种平台产生。这增加了批次效应的可能性。目前有几种批次评估方法,如主成分分析(PCA;主要基于视觉检查),但有时它们无法揭示所有潜在的批次效应。这些方法还可能导致无意中纠正归因于批次效应的生物学上有趣的因素的风险。在这里,我们提出了一种新的统计方法,基于概率主成分和协变量分析(PPCCA)的发现批次效应(findBATCH)来评估批次效应。相同的框架还为批次校正提供了一种新方法,即校正批次效应(correctBATCH),我们已经证明这种方法比传统的基于 PCA 的校正方法更好。我们通过合并来自不同研究的基因表达数据,在诊断和校正批次效应并保留生物学效应后,使用两个不同的示例(乳腺癌和结直肠癌)展示了这些方法的实用性。这些方法与基于传统视觉检查的 PCA 一起,可作为探索批次效应(exploBATCH;https://github.com/syspremed/exploBATCH)的 R 包的一部分。