Academy of Pharmacy, Xi'an Jiaotong-Liverpool University, 111 Ren'ai Road, Dushu Lake Higher Education Town, Suzhou Industrial Park, Suzhou 215123, Jiangsu Province, PRC.
Clinical Bioinformatics, Gilead Sciences, Inc., 333 Lakeside Dr, Foster City, CA 94404.
Biostatistics. 2023 Jul 14;24(3):635-652. doi: 10.1093/biostatistics/kxab039.
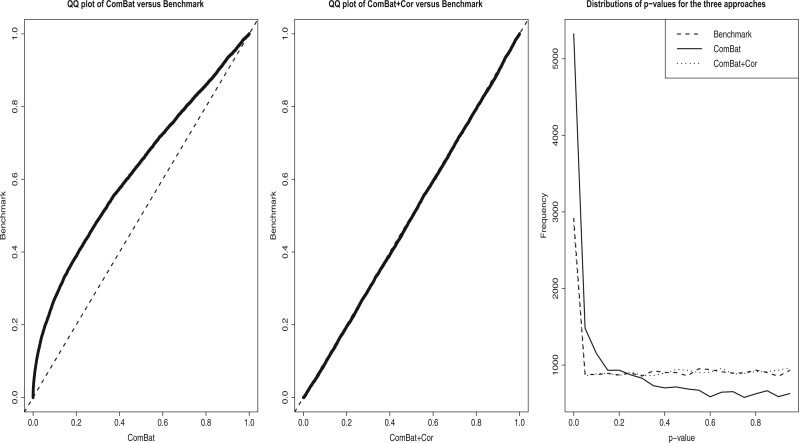
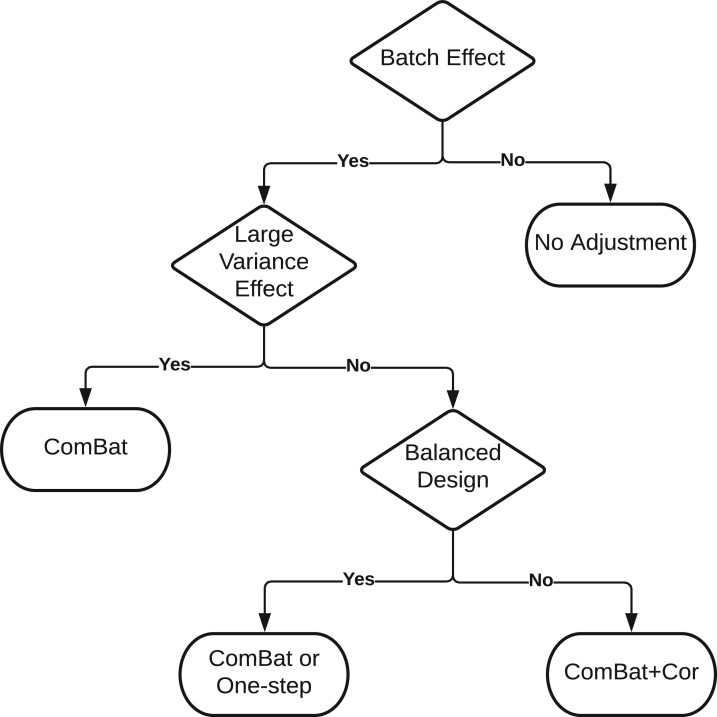
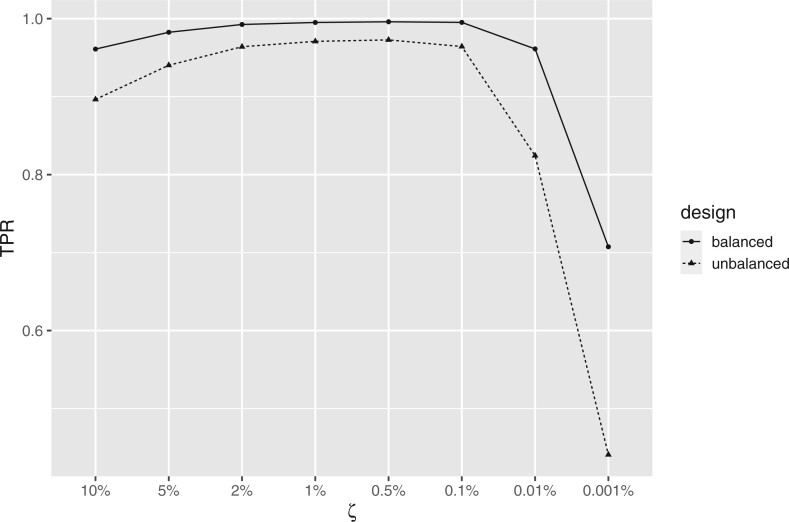
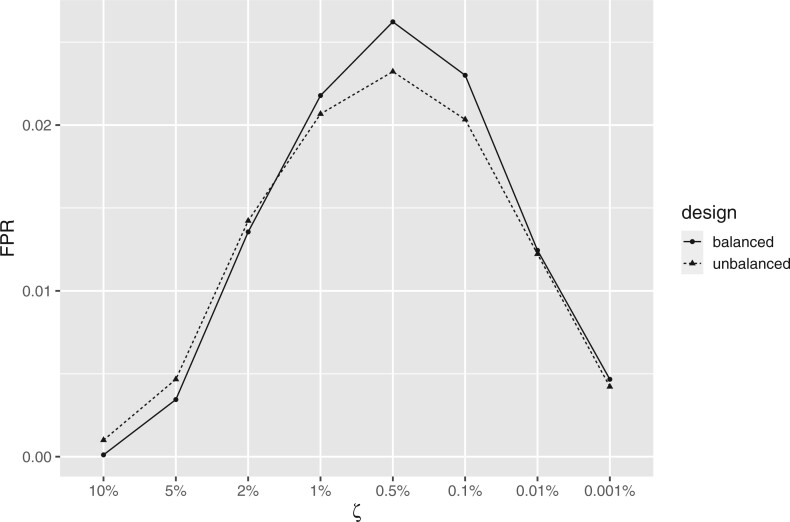
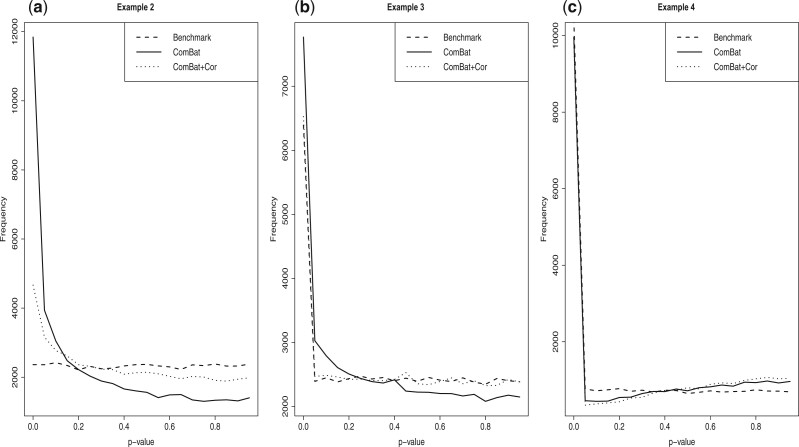
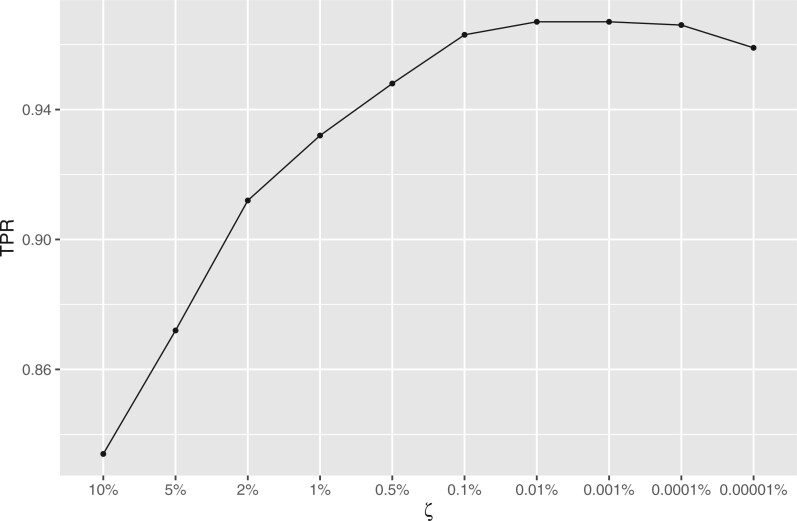
Nonignorable technical variation is commonly observed across data from multiple experimental runs, platforms, or studies. These so-called batch effects can lead to difficulty in merging data from multiple sources, as they can severely bias the outcome of the analysis. Many groups have developed approaches for removing batch effects from data, usually by accommodating batch variables into the analysis (one-step correction) or by preprocessing the data prior to the formal or final analysis (two-step correction). One-step correction is often desirable due it its simplicity, but its flexibility is limited and it can be difficult to include batch variables uniformly when an analysis has multiple stages. Two-step correction allows for richer models of batch mean and variance. However, prior investigation has indicated that two-step correction can lead to incorrect statistical inference in downstream analysis. Generally speaking, two-step approaches introduce a correlation structure in the corrected data, which, if ignored, may lead to either exaggerated or diminished significance in downstream applications such as differential expression analysis. Here, we provide more intuitive and more formal evaluations of the impacts of two-step batch correction compared to existing literature. We demonstrate that the undesired impacts of two-step correction (exaggerated or diminished significance) depend on both the nature of the study design and the batch effects. We also provide strategies for overcoming these negative impacts in downstream analyses using the estimated correlation matrix of the corrected data. We compare the results of our proposed workflow with the results from other published one-step and two-step methods and show that our methods lead to more consistent false discovery controls and power of detection across a variety of batch effect scenarios. Software for our method is available through GitHub (https://github.com/jtleek/sva-devel) and will be available in future versions of the $\texttt{sva}$ R package in the Bioconductor project (https://bioconductor.org/packages/release/bioc/html/sva.html).
非可忽略的技术变异在来自多个实验运行、平台或研究的数据中通常是观察到的。这些所谓的批次效应可能导致难以合并来自多个来源的数据,因为它们会严重偏向分析的结果。许多研究小组已经开发了从数据中去除批次效应的方法,通常是通过将批次变量纳入分析(一步校正)或在正式或最终分析之前对数据进行预处理(两步校正)。由于其简单性,一步校正通常是可取的,但它的灵活性有限,并且在分析具有多个阶段时,很难统一包含批次变量。两步校正允许更丰富的批次均值和方差模型。然而,先前的研究表明,两步校正可能导致下游分析中的不正确统计推断。一般来说,两步方法在校正后的数据中引入了相关结构,如果忽略了这种结构,可能会导致下游应用(如差异表达分析)中的显著性被夸大或减弱。在这里,我们提供了比现有文献更直观和更正式的两步批量校正影响的评估。我们证明了两步校正的不良影响(夸大或减弱显著性)取决于研究设计的性质和批次效应。我们还提供了在下游分析中使用校正后数据的估计相关矩阵克服这些负面影响的策略。我们比较了我们提出的工作流程的结果与其他已发表的一步和两步方法的结果,并表明我们的方法在各种批次效应情况下导致更一致的错误发现控制和检测能力。我们的方法的软件可通过 GitHub(https://github.com/jtleek/sva-devel)获得,并将在 Bioconductor 项目中的未来版本的 $\texttt{sva}$ R 包中提供(https://bioconductor.org/packages/release/bioc/html/sva.html)。